.svg) National Institute of General Medical Sciences |
 |
|
National Biomedical Resource for |
| ACERT's Service and Collaborative Projects | |||||||||
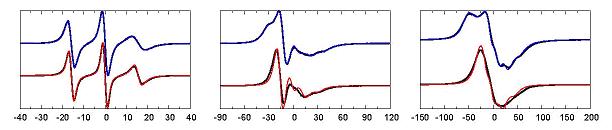
ESR spectra are rich in information that can be related to the structure and function of the spin labeled bimolecule. Nonetheless, inferring molecular detail from the spectra is difficult due to the complexity introduced by the internal dynamics of the spin-label, such as the commonly used R1. Our molecular dynamics(MD) simulations of a fully solvated, poly-alanine α-helix containing one spin labeled (R1) cysteine residue at its central position was chosen as an idealized model of R1 at a solvent-exposed helix surface site in proteins. Due to the relatively small size of the system, we were able to simulate 18 independent trajectories, each extending for 100 ns. In spite of the reasonably long duration of the simulations, each individual trajectory failed to exhaustively sample all the conformations that were accessible to the spin label. As a result, the times that the R1 was observed to spend in its various conformations do not necessarily reflect the correct state probabilities, but likely depend on the starting conformations. On the other hand, when taken together, the trajectories seemed to explore a significant realm of conformational possibilities. The issue begs for a robust analysis method to extract this information from a collection of MD trajectories. One such approach would be to relate the MD results to a Markov jump process. The main idea is that many independent trajectories can be used to estimate conditional transition probabilities, even though each trajectory does not necessarily reflect the correct equilibrium probabilities. To this end, the detailed dynamics of the MD trajectories has to be mapped to a discrete-state Markov jump model, the state-to-state transition probability matrix of which needs to be determined. Once its parameters have been properly estimated, the so-constructed Markov model should allow for the generation of arbitrarily long stochastic trajectories, which can then be used to simulate ESR spectra in the time domain. We have developed a procedure that constructs a Markov model with the desired separation of time scales from the MD trajectories. ESR spectra at three different frequencies were simulated from the trajectories generated by this model and compared favorably with spectra simulated directly from the MD trajectories. (cf. Figure: the red and blue spectra result from two rarely interconverting sets of Markov states.) This study shows that such a procedure holds the potential of bridging the gap between atomistic MD simulations of solvated spin labeled proteins and their experimental ESR spectra. Publication: D. Sezer, J.H. Freed, and B. Roux, J. Phys. Chem. B, 112, 11014-11027 (2008); PMC2562300 |
|||||||||
|
|||||||||
|
D. Sezer, (Department of Physics and Department of Biochemistry and Molecular Biology, The University of Chicago, Chicago, Illinois) J. H. Freed, (ACERT) B. Roux (Department of Biochemistry and Molecular Biology, The University of Chicago, Chicago, Illinois) |
|||||||||
|
|||||||||
|
About ACERT Contact Us |
Research |
Outreach |
ACERT is supported by grant 1R24GM146107 from the National Institute of General Medical Sciences (NIGMS), part of the National Institutes of Health. |
|||||
| ||||||||