.svg) National Institute of General Medical Sciences |
 |
|
National Biomedical Resource for |
| Theory and computational methods |
|
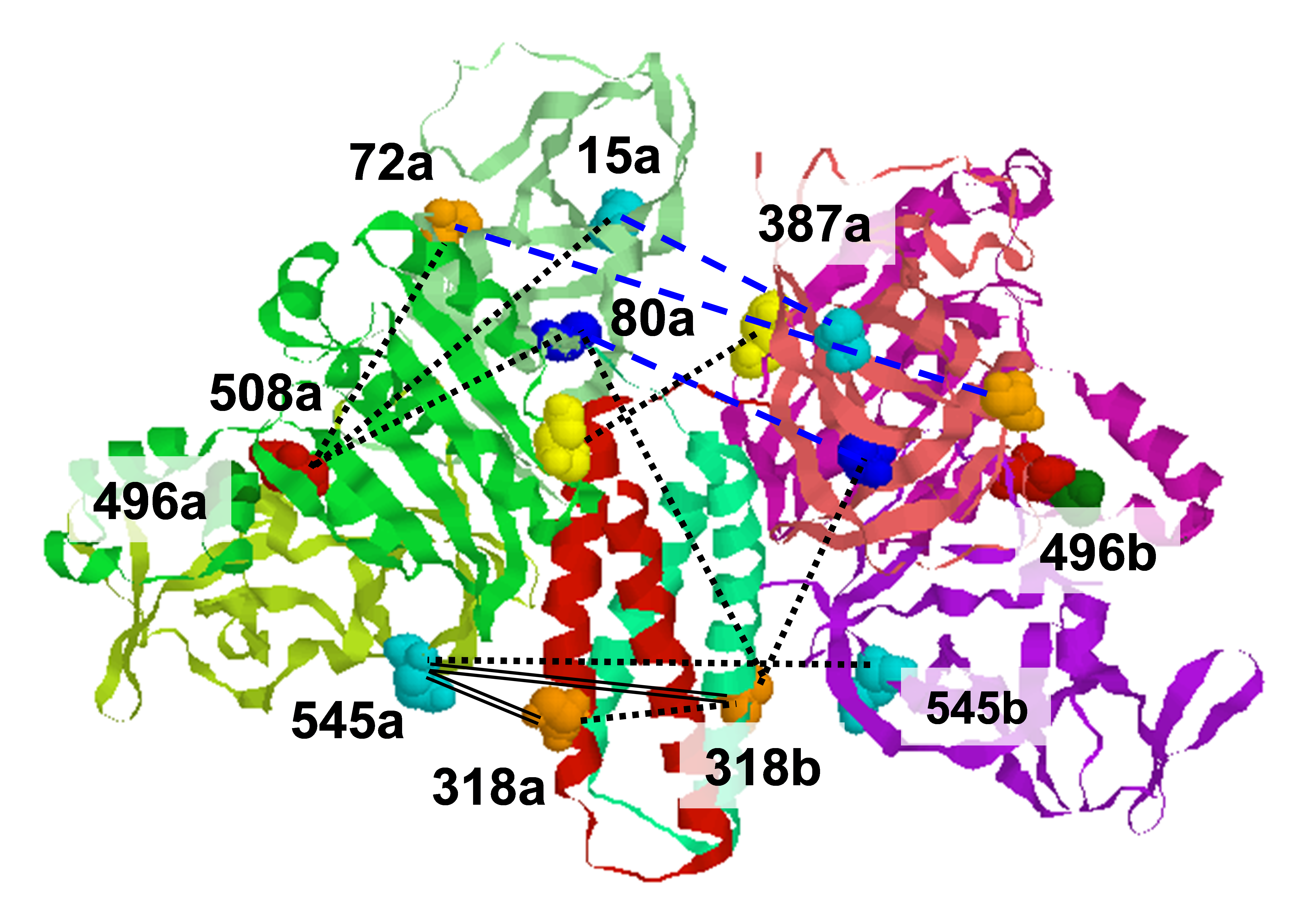
The overall aim of ACERT in this area is to further develop the computational methods needed to interpret multi-frequency cw and 2D-ELDOR spectra in terms of powerful theoretical methods based on the stochastic Liouville equation and on molecular dynamics simulations in support of the extensive experimental projects. The experimentally observed magnetic resonance lineshape is the result of the detailed structure and motion of the spin labeled molecules and, indirectly, of the surroundings. Roughly speaking, the magnetic tensors of the spin label determine the resonant frequencies for a given magnetic field strength, while the motion of the labeled molecules and its surroundings gives rise to time dependent fluctuations in the spin energy levels and concomitant loss of phase coherence (T2 processes) as well as energy dissipation (T1 processes) that are manifested, for example, as characteristic broadening and shifting of spectral features. If the motion of the labeled molecules is sufficiently fast, then the widths and positions of the peaks in the ESR spectrum can be calculated by simple perturbative methods. Simplistically speaking, in this regime the fluctuations in the orientation happen faster than the spin can “follow” so that the spin behaves as if it were evolving under the influence of an effective spin Hamiltonian which incorporates the effects of the rotational diffusion on the anisotropic part of the spin Hamiltonian in an “average” sense, also yielding dissipative terms that give rise to linewidths that decrease with increasing motional rates. Therefore, this fast motional regime is often referred to as the motional narrowing regime. The corresponding theory is well known, and the corresponding calculations involve explicit formulae for the first non-vanishing term in the perturbative expansion. However, the quantitative information available is limited to a handful of correlation times and parameters describing the equilibrium orientational distribution. Therefore, less emphasis will be placed on this so-called “fast motional regime”. The far more interesting and challenging case is the so-called “slow motional regime” where, in the presence of slower motions or stronger magnetic interactions due to use of higher magnetic fields, the dynamics of the spin system becomes intimately entangled with the molecular dynamics in a manner that is fundamentally inaccessible to perturbative methods. This situation is frequently the case for ESR, but very rarely for NMR. Not surprisingly, much more sophisticated theoretical and computational methods must be employed to calculate spectra in the slow-motional regime. The most effective computational methods developed to date for calculating slow-motional magnetic resonance lineshapes are based on discretization of the stochastic Liouville equation (SLE) using generalized spherical harmonic expansions. This approach has become the de facto standard because of the simplicity of evaluating matrix elements and the availability of extremely powerful iterative methods that exploit the symmetry and sparsity of the resulting matrix representation. In turn, the power and efficiency of these methods has made it possible to develop automated non-linear least-squares fitting procedures to identify optimal parameter sets for a wide variety of experiments. Extraction of quantitative information on molecular dynamics and ordering from slow-motional magnetic resonance spectra requires the use of detailed models of the dynamic structure of the system at multiple scales of time and length. In membranes for example, the experimentally observed spectrum of a spin-labeled molecule is affected by microscopic parameters such as the magnetic tensors, quantities such as the diffusion tensor that inter-relate microscopic and mesoscopic regimes (moments of inertia and rotational viscosities, respectively), the mesoscopic orientational potential exerted by the local liquid crystalline ordering in the membrane, and macroscopic disorder resulting from variation in the local ordering throughout the sample. From this statement alone, one may conclude that numerical calculation of magnetic resonance spectra that can be directly compared to experimental results is an extremely difficult problem requiring a comprehensive theoretical framework, sophisticated numerical techniques, and powerful computers. |
|
© 2022 |
|
About ACERT Contact Us |
Research |
Outreach |
ACERT is supported by grant 1R24GM146107 from the National Institute of General Medical Sciences (NIGMS), part of the National Institutes of Health. |
|||||
| ||||||||
