|
MRI Denoising Using Pixel-Wise Threshold Selection
N. Srivastava, G.R. Sahoo, H. U. Voss, S. N. Niogi, J. H. Freed, and M. Srivastava
IEEE Access (In press)
|
|
|
MRI Denoising Using Pixel-Wise Threshold Selection
N. Srivastava, G.R. Sahoo, H. U. Voss, S. N. Niogi, J. H. Freed, and M. Srivastava
IEEE Access (In press)
|
|
|
|
|
|
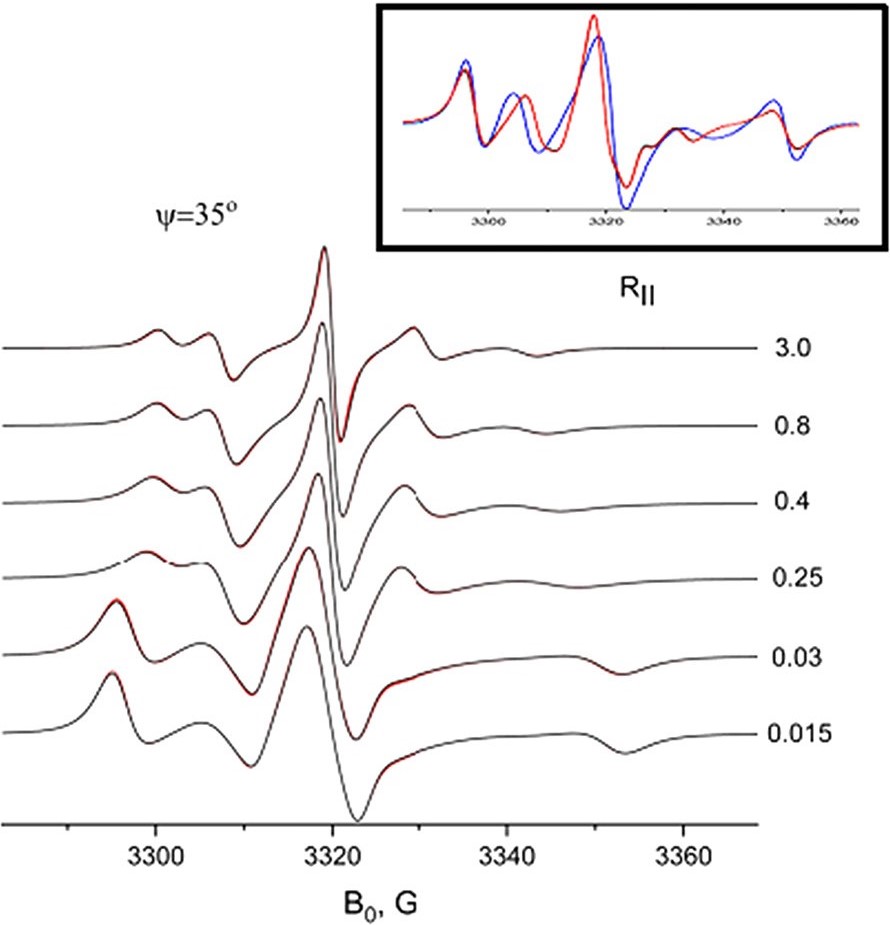
An analysis of double-quantum coherence ESR in an N-spin system: Analytical expressions and predictions
A. Sinha Roy, J. A. Marohn, J. H. Freed
J. Chem. Phys. 160, 134105 (2024)
|
|
|
An analysis of double-quantum coherence ESR in an N-spin system: Analytical expressions and predictions
A. Sinha Roy, J. A. Marohn, J. H. Freed
J. Chem. Phys. 160, 134105 (2024)
<doi: 10.1063/5.0200054>
PMID:
38557852
PMCID:
PMC11087869
|
|
|
ABSTRACT: Electron spin resonance pulsed dipolar spectroscopy (PDS) has become popular in protein 3D structure analysis. PDS studies yield distance distributions between a pair or multiple pairs of spin probes attached to protein molecules, which can be used directly in structural studies or as constraints in theoretical predictions. Double-quantum coherence (DQC) is a highly sensitive and accurate PDS technique to study protein structures in the solid state and under physiologically relevant conditions. In this work, we have derived analytical expressions for the DQC signal for a system with N-dipolar coupled spin-1/2 particles in the solid state. The expressions are integrated over the relevant spatial parameters to obtain closed form DQC signal expressions. These expressions contain the concentration-dependent "instantaneous diffusion" and the background signal. For micromolar and lower concentrations, these effects are negligible. An approximate analysis is provided for cases of finite pulses. The expressions obtained in this work should improve the analysis of DQC experimental data significantly, and the analytical approach could be extended easily to a wide range of magnetic resonance phenomena.
|
|
|
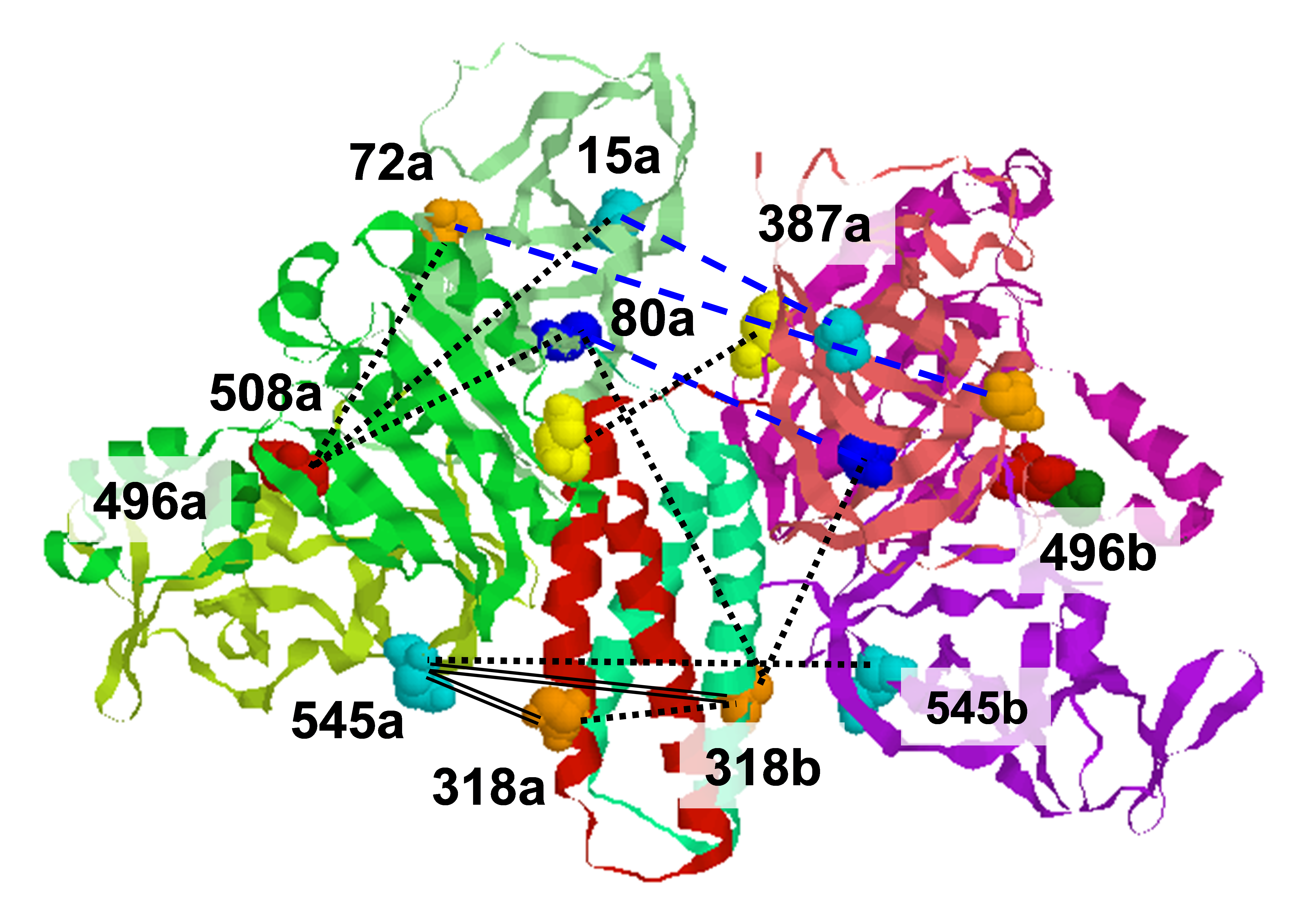
The crystal structure of bacteriophage λ RexA provides novel insights into the DNA binding properties of Rex-like phage exclusion proteins
M. C. Adams, C. J. Schiltz, J. Sun, C. J. Hosford, V. M. Johnson, H. Pan, P. P. Borbat, J. H. Freed, L. C. Thomason, C. Court, D. L. Court, J. S. Chappie
Nucleic Acids Res. 52, 4659-4675 (2024)
|

|
|
The crystal structure of bacteriophage λ RexA provides novel insights into the DNA binding properties of Rex-like phage exclusion proteins
M. C. Adams, C. J. Schiltz, J. Sun, C. J. Hosford, V. M. Johnson, H. Pan, P. P. Borbat, J. H. Freed, L. C. Thomason, C. Court, D. L. Court, J. S. Chappie
Nucleic Acids Res. 52, 4659-4675 (2024)
Supporting Information
<doi: 10.1093/nar/gkae212>
PMID:
38554102
PMCID:
PMC11077077
|
|
|
ABSTRACT: RexA and RexB function as an exclusion system that prevents bacteriophage T4rII mutants from growing on Escherichia coli λ phage lysogens. Recent data established that RexA is a non-specific DNA binding protein that can act independently of RexB to bias the λ bistable switch toward the lytic state, preventing conversion back to lysogeny. The molecular interactions underlying these activities are unknown, owing in part to a dearth of structural information. Here, we present the 2.05-Å crystal structure of the λ RexA dimer, which reveals a two-domain architecture with unexpected structural homology to the recombination-associated protein RdgC. Modelling suggests that our structure adopts a closed conformation and would require significant domain rearrangements to facilitate DNA binding. Mutagenesis coupled with electromobility shift assays, limited proteolysis, and double electron–electron spin resonance spectroscopy support a DNA-dependent conformational change. In vivo phenotypes of RexA mutants suggest that DNA binding is not a strict requirement for phage exclusion but may directly contribute to modulation of the bistable switch. We further demonstrate that RexA homologs from other temperate phages also dimerize and bind DNA in vitro. Collectively, these findings advance our mechanistic understanding of Rex functions and provide new evolutionary insights into different aspects of phage biology.
|
|
|
Phosphorylation, disorder, and phase separation govern the behavior of Frequency in the fungal circadian clock
D. Tariq, N. Maurici, B. M. Bartholomai, S. Chandrasekaran, J. C. Dunlap, A. Bah, B. R. Crane
eLife 12 RP90259 (2024)
Supporting Information
<doi: 10.7554/eLife.90259>
PMID:
38526948
PMCID:
PMC10963029
|

|
|
ABSTRACT: Circadian clocks are composed of transcription-translation negative feedback loops that pace rhythms of gene expression to the diurnal cycle. In the filamentous fungus Neurospora crassa, the proteins Frequency (FRQ), the FRQ-interacting RNA helicase (FRH), and Casein-Kinase I (CK1) form the FFC complex that represses expression of genes activated by the white-collar complex (WCC). FRQ orchestrates key molecular interactions of the clock despite containing little predicted tertiary structure. Spin labeling and pulse-dipolar electron spin resonance spectroscopy provide domain-specific structural insights into the 989-residue intrinsically disordered FRQ and the FFC. FRQ contains a compact core that associates and organizes FRH and CK1 to coordinate their roles in WCC repression. FRQ phosphorylation increases conformational flexibility and alters oligomeric state, but the changes in structure and dynamics are non-uniform. Full-length FRQ undergoes liquid–liquid phase separation (LLPS) to sequester FRH and CK1 and influence CK1 enzymatic activity. Although FRQ phosphorylation favors LLPS, LLPS feeds back to reduce FRQ phosphorylation by CK1 at higher temperatures. Live imaging of Neurospora hyphae reveals FRQ foci characteristic of condensates near the nuclear periphery. Analogous clock repressor proteins in higher organisms share little position-specific sequence identity with FRQ; yet, they contain amino acid compositions that promote LLPS. Hence, condensate formation may be a conserved feature of eukaryotic clocks.
|
|
|
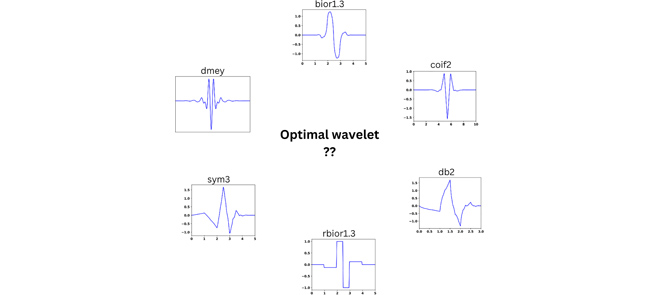
Optimal Wavelet Selection for Signal Denoising
G. R. Sahoo, J. H. Freed, M. Srivastava
IEEE Access 12 45369-45380 (2024)
|
|
|
Optimal Wavelet Selection for Signal Denoising
G. R. Sahoo, J. H. Freed, M. Srivastava
IEEE Access 12 45369-45380 (2024)
<doi: 10.1109/ACCESS.2024.3377664>
PMID: [In Progress] PMCID:
[In Progress]
|
|
|
ABSTRACT: Wavelet denoising plays a key role in removing noise from signals and is widely used in many applications. In denoising, selection of the mother wavelet is desirable for maximizing the separation of noise and signal coefficients in the wavelet domain for effective noise thresholding. At present, wavelet selection is carried out in a heuristic manner or using a trial-and-error that is time consuming and prone to error, including human bias. This paper introduces a universal method to select optimal wavelets based on the sparsity of Detail components in the wavelet domain, an empirical approach. A mean of sparsity change ( μsc ) parameter is defined that captures the mean variation of noisy Detail components. The efficacy of the presented method is tested on simulated and experimental signals from Electron Spin Resonance spectroscopy at various SNRs. The results reveal that the μsc values of signal vary abruptly between wavelets, whereas for noise it displays similar values for all wavelets. For low Signal-to-Noise Ratio (SNR) data, the change in μsc between highest and second highest value is ≈8–10% and for high SNR data it is around 5%. The mean of sparsity change increases with the SNR of the signal, which implies that multiple wavelets can be used for denoising a signal, whereas, the signal with low SNR can only be efficiently denoised with a few wavelets. Either a single wavelet or a collection of optimal wavelets (i.e., top five wavelets) should be selected from the highest μsc values. The code is available on GitHub and the signalsciencelab.com website.
|
|
|
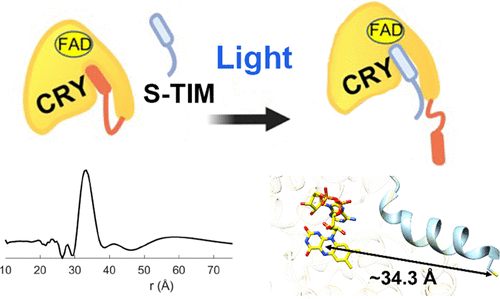
Dissecting the Interaction between Cryptochrome and Timeless Reveals Underpinnings of Light-Dependent Recognition
C. M. Schneps, R. Dunleavy, B. R. Crane
Biochemistry 63 Online ahead of print (2024)
|
|
|
Dissecting the Interaction between Cryptochrome and Timeless Reveals Underpinnings of Light-Dependent Recognition
C. M. Schneps, R. Dunleavy, B. R. Crane
Biochemistry 63 Online ahead of print (2024)
<doi: 10.1021/acs.biochem.3c00630>
PMID:
38294880
PMCID:
PMC11289166
|
|
|
ABSTRACT: Circadian rhythms are determined by cell-autonomous transcription–translation feedback loops that entrain to environmental stimuli. In the model circadian clock of Drosophila melanogaster, the clock is set by the light-induced degradation of the core oscillator protein timeless (TIM) by the principal light-sensor cryptochrome (CRY). The cryo-EM structure of CRY bound to TIM revealed that within the extensive CRY:TIM interface, the TIM N-terminus binds into the CRY FAD pocket, in which FAD and the associated phosphate-binding loop (PBL) undergo substantial rearrangement. The TIM N-terminus involved in CRY binding varies in isoforms that facilitate the adaptation of flies to different light environments. Herein, we demonstrate, through peptide binding assays and pulsed-dipolar electron spin resonance (ESR) spectroscopy, that the TIM N-terminal peptide alone exhibits light-dependent binding to CRY and that the affinity of the interaction depends on the initiating methionine residue. Extensions to the TIM N-terminus that mimic less light-sensitive variants have substantially reduced interactions with CRY. Substitutions of CRY residues that couple to the flavin rearrangement in the CRY:TIM complex have dramatic effects on CRY light activation. CRY residues Arg237 on α8, Asn253, and Gln254 on the PBL are critical for the release of the CRY autoinhibitory C-terminal tail (CTT) and subsequent TIM binding. These key light-responsive elements of CRY are well conserved throughout Type I cryptochromes of invertebrates but not by cryptochromes of chordates and plants, which likely utilize a distinct light-activation mechanism.
|
|
|
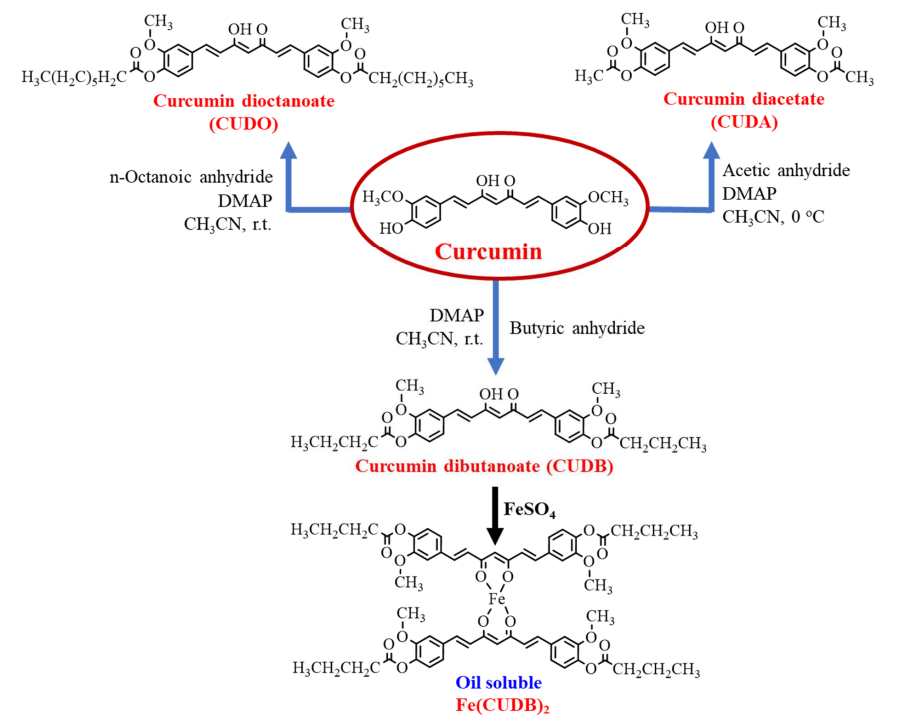
Oil soluble iron: Curcumin derivatives and their complex
A. Zarei, L. Khazdooz, A. Khojastegi, A. A. Altaf, A. Abbaspourrad
Food Chem. 431 137085 (2024)
|
|
|
Oil soluble iron: Curcumin derivatives and their complex
A. Zarei, L. Khazdooz, A. Khojastegi, A. A. Altaf, A. Abbaspourrad
Food Chem. 431 137085 (2024)
<doi: 10.1016/j.foodchem.2023.137085>
PMID:
37567079
PMCID:
PMC10566601
|
|
|
ABSTRACT: Curcumin dibutanoate (CUDB) is a new oil soluble bidentate ligand which shows higher stability against heat and oxidation compared to curcumin. The oil solubility of this ligand increased an order of magnitude over curcumin. This biomolecule showed high digestibility in a simulated intestinal trial and was hydrolyzed in the presence of porcine pancreatin releasing ∼91% of the curcumin. When curcumin dibutanoate was complexed with Fe2+, Fe(CUDB)2 was formed as a new iron (II) complex. Due to the high hydrophobicity of the curcumin dibutanoate ligand, the solubility of Fe(CUDB)2 was found to be 2.8 mg/mL in canola oil. The steric hindrance afforded by the CUDB ligand, coupled with its hydrophobicity stabilized the iron (II) oxidation state within the complex compared to FeSO4⋅7H2O as measured by 2,2-diphenyl-1-picrylhydrazyl (DPPH) radical. Fe(CUDB)2 has potential to be a new form of oil-soluble iron supplement which co-delivers iron (II) and curcumin.
|
|
|
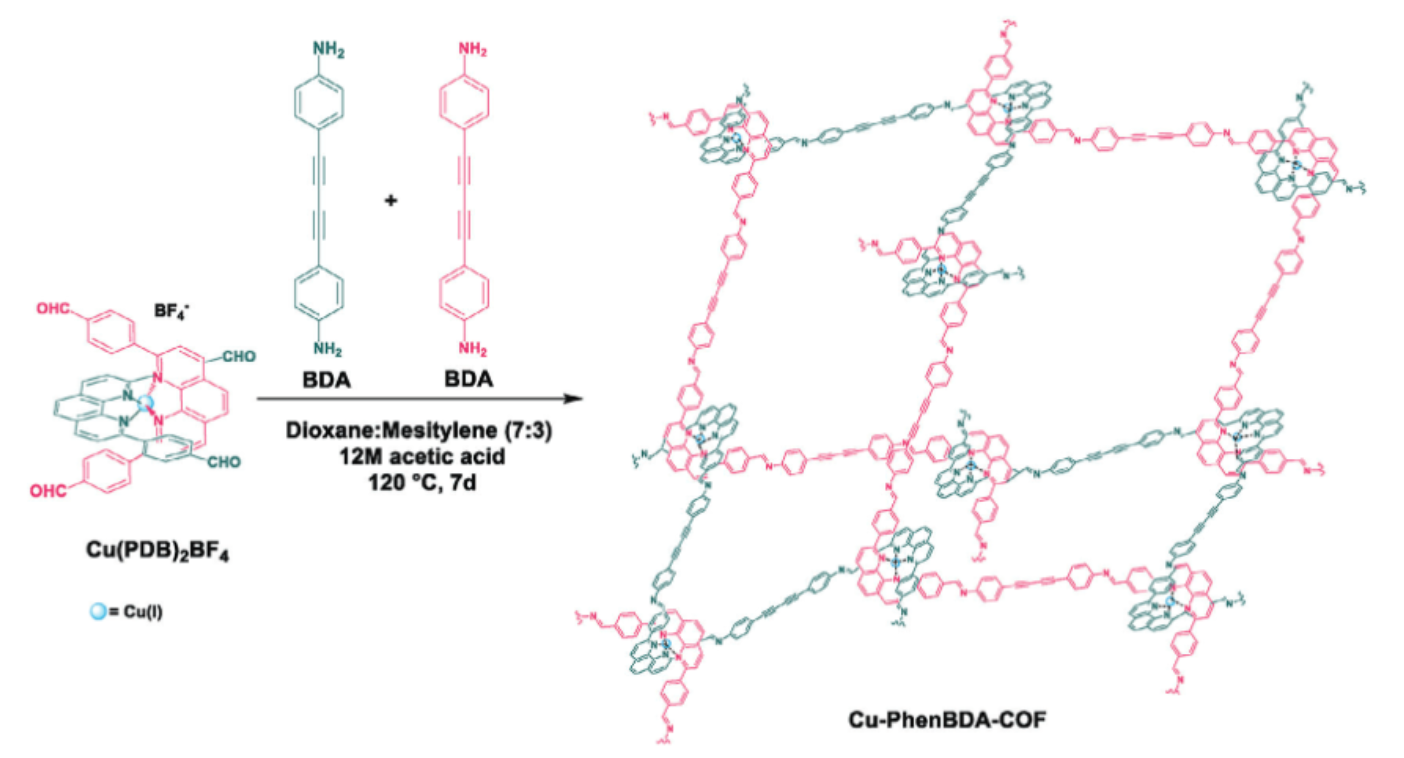
Modulating Narrow Bandgap in a Diacetylene Functionalized Woven Covalent Organic Framework as a Visible Light Responsive Photocatalyst
A. Khojastegi, A. Khosropour, S. Amirjalayer, I. Mosleh, A. Abbaspourrad
Adv. Funct. Mater. 34, 2309367 (2023)
|
|
|
Modulating Narrow Bandgap in a Diacetylene Functionalized Woven Covalent Organic Framework as a Visible Light Responsive Photocatalyst
A. Khojastegi, A. Khosropour, S. Amirjalayer, I. Mosleh, A. Abbaspourrad
Adv. Funct. Mater. 34, 2309367 (2023)
<doi: 10.1002/adfm.202309367>
PMID: [In Progress] PMCID:
[In Progress]
|
|
|
ABSTRACT: Woven covalent organic frameworks (COF) possess entangled 3D frameworks. The metallated version of these structures contains spatially isolated Cu(I) centers and promising optoelectronic properties because of metal-to-ligand charge transfer (MLCT). However, despite their potential, woven COFs have not yet been investigated as photocatalysts. In this study, a new woven COF, Cu-PhenBDA-COF, functionalized with diacetylene bonds is developed. Cu-PhenBDA-COF is fully characterized, and the optoelectronic and photocatalytic properties are compared to previously reported Cu-COF-505. The diacetylene bonds of the linker positively impact the optoelectronic properties of Cu-PhenBDA-COF and result in a narrower bandgap and better charge separation efficiency. When the Cu(I) center is removed from both woven COFs, the absorption edge is blueshifted, resulting in a wider bandgap, and there is a considerable decrease in the charge separation efficiency, underscoring the pivotal role of MLCT. This trend is reflected in the photocatalytic activity of the woven COFs toward the degradation of sulfamethoxazole in water, where the highest reaction rate constant (kapp) is recorded for the metallated diacetylene functionalized woven COF, Cu-PhenBDA-COF.
|
|
|
Enzymatic Spin-Labeling of Protein N- and C-Termini for Electron Paramagnetic Resonance Spectroscopy
R. Dunleavy, S. Chandrasekaran, and B. R. Crane
Bioconj. Chem. 34, Online ahead of print (2023)
Supporting Information
<doi: 10.1021/acs.bioconjchem.3c00029>
PMID:
36921260
PMCID:
PMC10502183
|

|
|
ABSTRACT: Electron paramagnetic resonance (EPR) spectroscopy is a powerful tool for investigating the structure and dynamics of proteins. The introduction of paramagnetic moieties at specific positions in a protein enables precise measurement of local structure and dynamics. This technique, termed site-directed spin-labeling, has traditionally been performed using cysteine-reactive radical-containing probes. However, large proteins are more likely to contain multiple cysteine residues and cysteine labeling at specific sites may be infeasible or impede function. To address this concern, we applied three peptide-ligating enzymes (sortase, asparaginyl endopeptidase, and inteins) for nitroxide labeling of N- and C-termini of select monomeric and dimeric proteins. Continuous wave and pulsed EPR (double electron electron resonance) experiments reveal specific attachment of nitroxide probes to ether N-termini (OaAEP1) or C-termini (sortase and intein) across three test proteins (CheY, CheA, and iLOV), thereby enabling a straightforward, highly specific, and general method for protein labeling. Importantly, the linker length (3, 5, and 9 residues for OaAEP1, intein, and sortase reactions, respectively) between the probe and the target protein has a large impact on the utility of distance measurements by pulsed EPR, with longer linkers leading to broader distributions. As these methods are only dependent on accessible N- and C-termini, we anticipate application to a wide range of protein targets for biomolecular EPR spectroscopy.
|
|
|
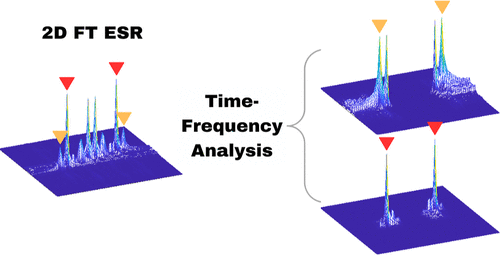
Time-Frequency Analysis of Two-Dimensional Electron Spin Resonance Signals
G. R. Sahoo, A. Sinha Roy, M. Srivastava
J. Phys. Chem. A 127, 7793-7801 (2023)
Supporting Information
<doi: 10.1021/acs.jpca.3c02708>
PMID:
37699569
PMCID:
PMC10529365
|
|
|
ABSTRACT: Two-dimensional electron spin resonance (2D ESR) spectroscopy is a unique experimental technique for probing protein structure and dynamics, including processes that occur at the microsecond time scale. While it provides significant resolution enhancement over the one-dimensional experimental setup, spectral broadening and noise make extraction of spectral information highly challenging. Traditionally, two-dimensional Fourier transform (2D FT) is applied for the analysis of 2D ESR signals, although its efficiency is limited to stationary signals. In addition, it often fails to resolve overlapping peaks in 2D ESR. In this work, we propose a time-frequency analysis of 2D time-domain signals, which identifies all frequency peaks by decoupling a signal into its distinct constituent components via projection on the time-frequency plane. The method utilizes 2D undecimated discrete wavelet transform (2D UDWT) as an intermediate step in the analysis, followed by signal reconstruction and 2D FT. We have applied the method to a simulated 2D double quantum coherence (DQC) signal for validation and a set of experimental 2D ESR signals, demonstrating its efficiency in resolving overlapping peaks in the frequency domain, while displaying frequency evolution with time in case of non-stationary data.
|
|
|
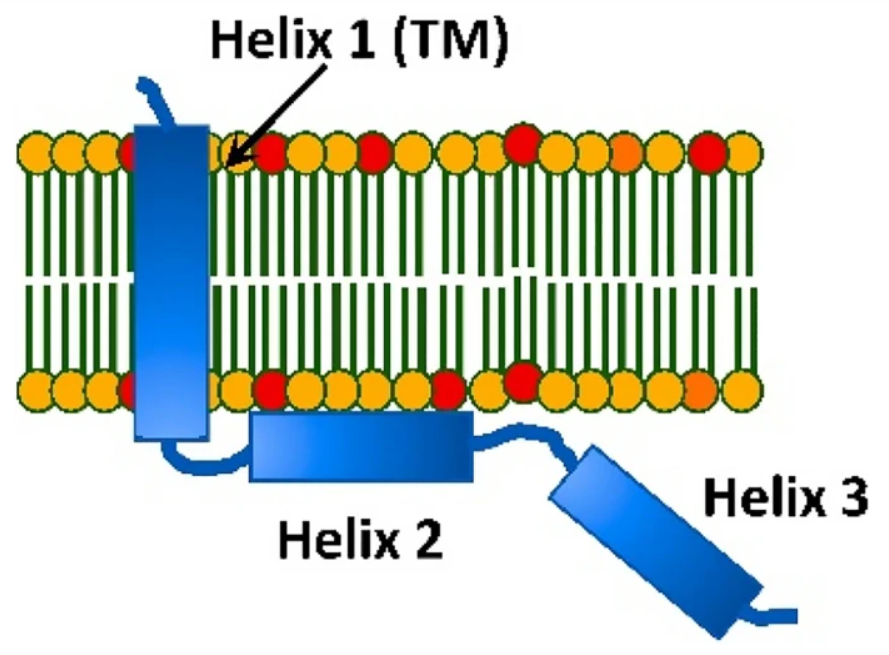
HIV-1 Vpu protein forms stable oligomers in aqueous solution via its transmembrane domain self-association
S. Majeed, L. Dang, M. M. Islam, O. Ishola, P. P. Borbat, S. J. Ludtke, and E. R. Georgieva
Scientific Reports 13, 14691 (2023)
Supporting Information
<doi: 10.1038/s41598-023-41873-0>
PMID:
37673923
PMCID:
PMC10483038
|
|
|
ABSTRACT: We report our findings on the assembly of the HIV-1 protein Vpu into soluble oligomers. Vpu is a key HIV-1 protein. It has been considered exclusively a single-pass membrane protein. Previous observations show that this protein forms stable oligomers in aqueous solution, but details about these oligomers still remain obscure. This is an interesting and rather unique observation, as the number of proteins transitioning between soluble and membrane embedded states is limited. In this study we made use of protein engineering, size exclusion chromatography, cryoEM and electron paramagnetic resonance (EPR) spectroscopy to better elucidate the nature of the soluble oligomers. We found that Vpu oligomerizes via its N-terminal transmembrane domain (TM). CryoEM suggests that the oligomeric state most likely is a hexamer/heptamer equilibrium. Both cryoEM and EPR suggest that, within the oligomer, the distal C-terminal region of Vpu is highly flexible. Our observations are consistent with both the concept of specific interactions among TM helices or the core of the oligomers being stabilized by hydrophobic forces. While this study does not resolve all of the questions about Vpu oligomers or their functional role in HIV-1 it provides new fundamental information about the size and nature of the oligomeric interactions.
|
|
|
Differentiating Unimodal and Multimodal Distributions in Pulsed Dipolar Spectroscopy Using Wavelet Transforms
A.Sinha Roy, J. H. Freed, M. Srivastava
Res. Sq. [Preprint] rs.3.rs-3216615 (2023)
Supporting Information
<doi: 10.21203/rs.3.rs-3216615/v1>
PMID:
37577617
PMCID:
PMC10418556
|
|
|
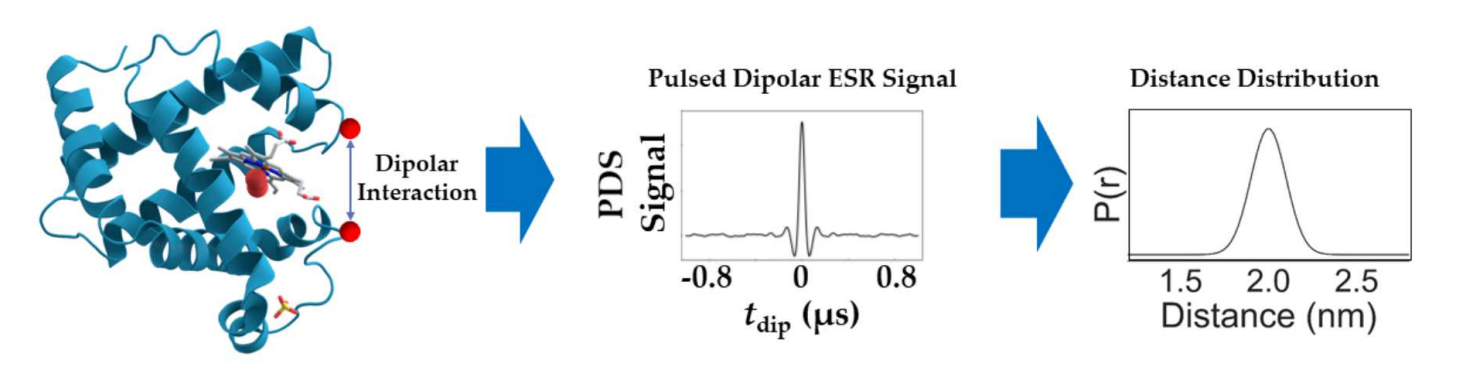
ABSTRACT: Site directed spin labeling has enabled protein structure determination using electron spin resonance (ESR) pulsed dipolar spectroscopy (PDS). Small details in a distance distribution can be key to understanding important protein structure-function relationships. A major challenge has been to differentiate unimodal and overlapped multimodal distance distributions. They often yield similar distributions and dipolar signals. Current model-free distance reconstruction techniques such as Srivastava-Freed Singular Value Decomposition (SF-SVD) and Tikhonov regularization can suppress these small features in uncertainty and/or error bounds, despite being present. In this work, we demonstrate that continuous wavelet transform (CWT) can distinguish PDS signals from unimodal and multimodal distance distributions. We show that periodicity in CWT representation reflects unimodal distributions, which is masked for multimodal cases. This work is meant as a precursor to a cross-validation technique, which could indicate the modality of the distance distribution.
|
|
|
Structural insights into perilipin 3 membrane association in response to diacylglycerol accumulation
Y. M. Choi, D. Ajjaji, K. D. Fleming, P. P. Borbat, M. L. Jenkins, B. E. Moeller, S. Fernando, S. R. Bhatia, J. H. Freed, J. E. Burke, A. R. Thiam, M. V. Airola
Nat. Commun. 14 3204 (2023)
|
|
|
Structural insights into perilipin 3 membrane association in response to diacylglycerol accumulation
Y. M. Choi, D. Ajjaji, K. D. Fleming, P. P. Borbat, M. L. Jenkins, B. E. Moeller, S. Fernando, S. R. Bhatia, J. H. Freed, J. E. Burke, A. R. Thiam, M. V. Airola
Nat. Commun. 14 3204 (2023)
Supporting Information
<doi: 10.1038/s41467-023-38725-w>
PMID:
37268630
PMCID:
PMC10238389
|
|
|
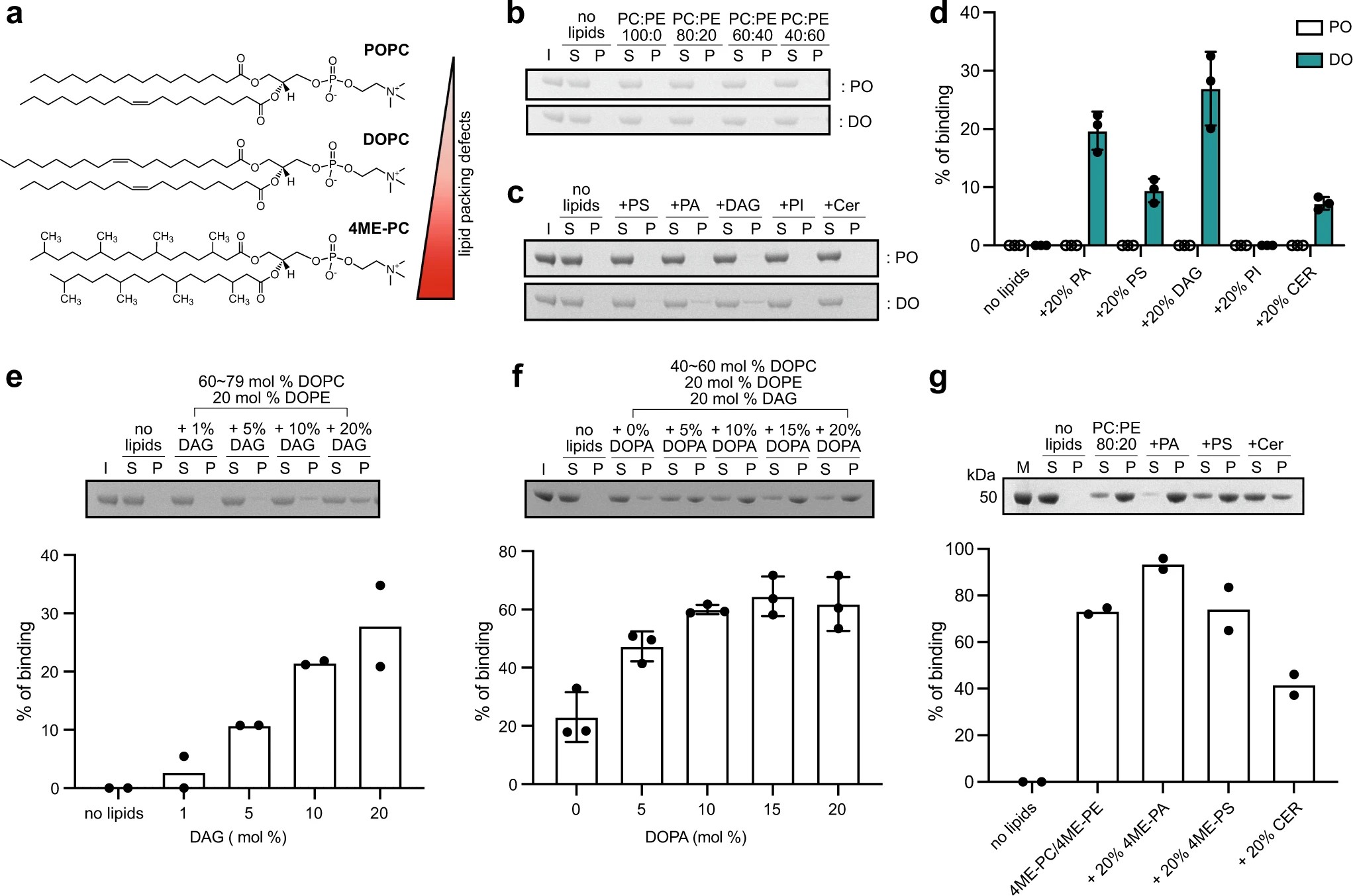
ABSTRACT: Lipid droplets (LDs) are dynamic organelles that contain an oil core mainly composed of triglycerides (TAG) that is surrounded by a phospholipid monolayer and LD-associated proteins called perilipins (PLINs). During LD biogenesis, perilipin 3 (PLIN3) is recruited to nascent LDs as they emerge from the endoplasmic reticulum. Here, we analyze how lipid composition affects PLIN3 recruitment to membrane bilayers and LDs, and the structural changes that occur upon membrane binding. We find that the TAG precursors phosphatidic acid and diacylglycerol (DAG) recruit PLIN3 to membrane bilayers and define an expanded Perilipin-ADRP-Tip47 (PAT) domain that preferentially binds DAG-enriched membranes. Membrane binding induces a disorder to order transition of alpha helices within the PAT domain and 11-mer repeats, with intramolecular distance measurements consistent with the expanded PAT domain adopting a folded but dynamic structure upon membrane binding. In cells, PLIN3 is recruited to DAG-enriched ER membranes, and this requires both the PAT domain and 11-mer repeats. This provides molecular details of PLIN3 recruitment to nascent LDs and identifies a function of the PAT domain of PLIN3 in DAG binding.
|
|
|
Thermal degradation of thaumatin at low pH and its prevention using alkyl gallates
B. Pomon, Y. Zhao, A. L. Lai, T. Lin, J. H. Freed, A. Abbaspourrad
Food Hydrocoll. 139, 108544 (2023)
Supporting Information
<doi: 10.1016/j.foodhyd.2023.108544>
PMID:
37546699
PMCID:
PMC10399911
|

|
|
ABSTRACT: Thaumatin, a potent sweet tasting protein extracted from the Katemfe Plant, is emerging as a natural alternative to synthetic non-nutritive sweeteners and flavor enhancer. As a food additive, its stability within the food matrix during thermal processing is of great interest to the food industry. When heated under neutral or basic conditions, thaumatin was found to lose its sweetness due to protein aggregation caused by sulfhydryl catalyzed disulfide bond interchange. At lower pH, while thaumatin was also found to lose sweetness after heating, it does so at a slower rate and shows more resistance to sweetness loss. SDS-PAGE indicated that thaumatin fragmented into multiple smaller pieces under heating in acidic pH. Using BEMPO-3, a lipophilic spin trap, we were able to detect the presence of a free-radical within the hydrophobic region of the protein during heating. Protein carbonyl content, a byproduct of protein oxidation, also increased upon heating, providing additional evidence for protein cleavage by a radical pathway. Hexyl gallate successfully inhibited the radical generation as well as protein carbonyl formation of thaumatin during heating.
|
|
|
A Simulation Independent Analysis of Single- and Multi-Component cw ESR Spectra
A. Sinha Roy, B. Dzikovski, D. Dolui, O. Makhlynets, A. Dutta, M. Srivastava
Magnetochemistry 9, 112 (2023)
Supporting Information
<doi: 10.3390/magnetochemistry9050112>
PMID:
37476293
PMCID:
PMC10357894
|
|
|
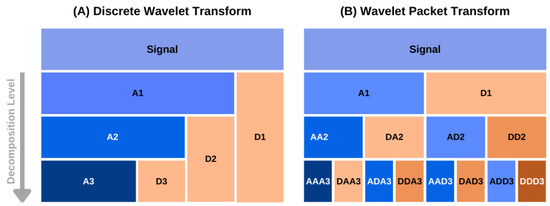
ABSTRACT: The accurate analysis of continuous-wave electron spin resonance (cw ESR) spectra of biological or organic free-radicals and paramagnetic metal complexes is key to understanding their structure–function relationships and electrochemical properties. The current methods of analysis based on simulations often fail to extract the spectral information accurately. In addition, such analyses are highly sensitive to spectral resolution and artifacts, users' defined input parameters and spectral complexity. We introduce a simulation-independent spectral analysis approach that enables broader application of ESR. We use a wavelet packet transform-based method for extracting g values and hyperfine (A) constants directly from cw ESR spectra. We show that our method overcomes the challenges associated with simulation-based methods for analyzing poorly/partially resolved and unresolved spectra, which is common in most cases. The accuracy and consistency of the method are demonstrated on a series of experimental spectra of organic radicals and copper–nitrogen complexes. We showed that for a two-component system, the method identifies their individual spectral features even at a relative concentration of 5% for the minor component.
|
|
|
Insights into the oligomeric structure of the HIV-1 Vpu protein
S. Majeed, O. Adetuyi, P. P. Borbat, M. M. Islam, O. Ishola, B. Zhao, E. R. Georgieva
J. Struct. Biol. 215, 107943 (2023)
Supporting Information
<doi: 10.1016/j.jsb.2023.107943>
PMID:
36796461
PMCID:
PMC10257199
|
|
|
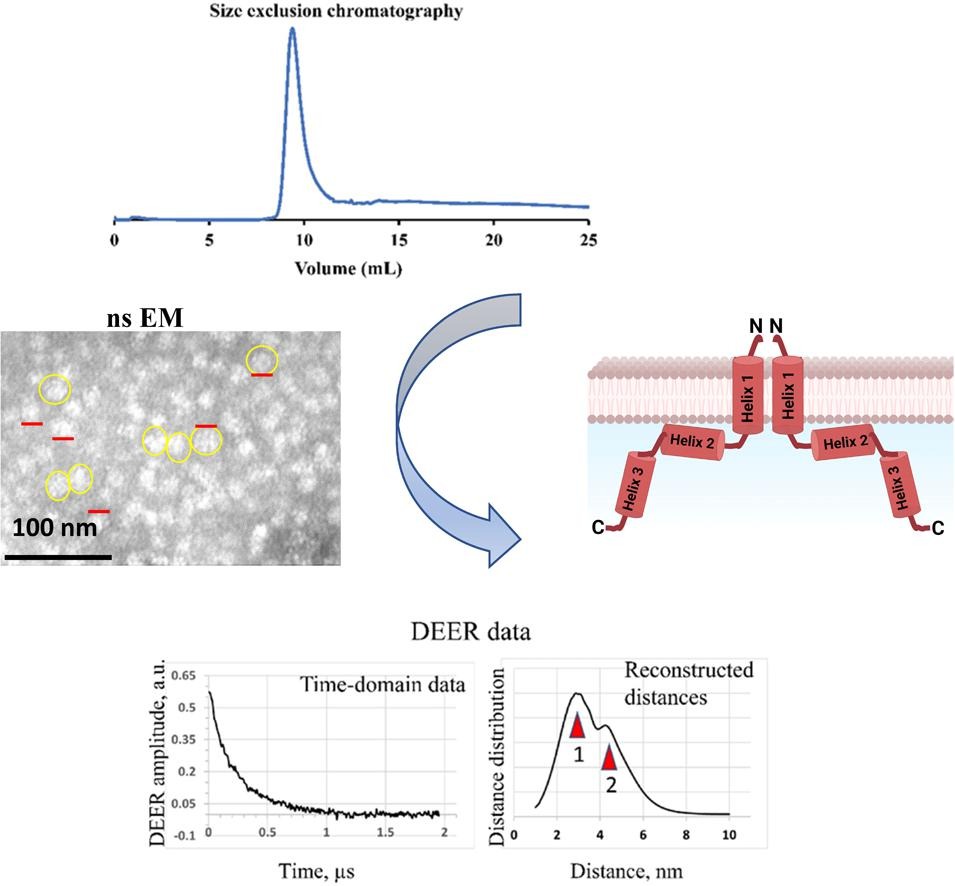
ABSTRACT: The HIV-1-encoded protein Vpu forms an oligomeric ion channel/pore in membranes and interacts with host proteins to support the virus lifecycle. However, Vpu molecular mechanisms are currently not well understood. Here, we report on the Vpu oligomeric organization under membrane and aqueous conditions and provide insights into how the Vpu environment affects the oligomer formation. For these studies, we designed a maltose-binding protein (MBP)-Vpu chimera protein and produced it in E. coli in soluble form. We analyzed this protein using analytical size-exclusion chromatography (SEC), negative staining electron microscopy (nsEM), and electron paramagnetic resonance (EPR) spectroscopy. Surprisingly, we found that MBP-Vpu formed stable oligomers in solution, seemingly driven by Vpu transmembrane domain self-association. A coarse modeling of nsEM data as well as SEC and EPR data suggests that these oligomers most likely are pentamers, similar to what was reported regarding membrane-bound Vpu. We also noticed reduced MBP-Vpu oligomer stability upon reconstitution of the protein in β-DDM detergent and mixtures of lyso-PC/PG or DHPC/DHPG. In these cases, we observed greater oligomer heterogeneity, with MBP-Vpu oligomeric order generally lower than in solution; however, larger oligomers were also present. Notably, we found that in lyso-PC/PG, above a certain protein concentration, MBP-Vpu assembles into extended structures, which had not been reported for Vpu. Therefore, we captured various Vpu oligomeric forms, which can shed light on Vpu quaternary organization. Our findings could be useful in understanding Vpu organization and function in cellular membranes and could provide information regarding the biophysical properties of single-pass transmembrane proteins.
|
|
|
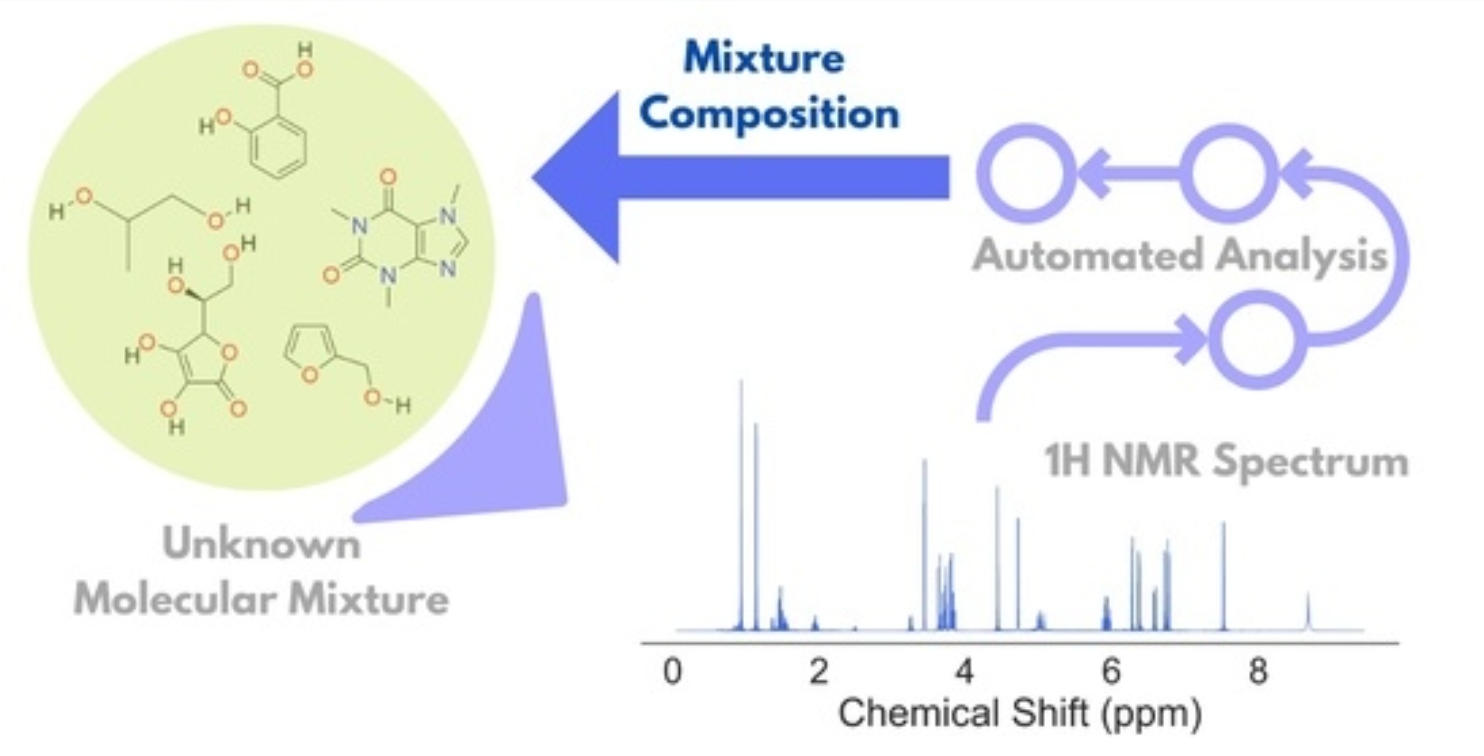
Unsupervised Analysis of Small Molecule Mixtures by Wavelet-Based Super-Resolved NMR
A. Sinha Roy, M. Srivastava
Molecules 28 792 (2023)
Supporting Information
<doi: 10.3390/molecules28020792>
PMID:
36677850
PMCID:
PMC9866129
|
|
|
ABSTRACT: Resolving small molecule mixtures by nuclear magnetic resonance (NMR) spectroscopy has been of great interest for a long time for its precision, reproducibility, and efficiency. However, spectral analyses for such mixtures are often highly challenging due to overlapping resonance lines and limited chemical shift windows. The existing experimental and theoretical methods to produce shift NMR spectra in dealing with the problem have limited applicability owing to sensitivity issues, inconsistency, and/or the requirement of prior knowledge. Recently, we resolved the problem by decoupling multiplet structures in NMR spectra by the wavelet packet transform (WPT) technique. In this work, we developed a scheme for deploying the method in generating highly resolved WPT NMR spectra and predicting the composition of the corresponding molecular mixtures from their 1H NMR spectra in an automated fashion. The four-step spectral analysis scheme consists of calculating the WPT spectrum, peak matching with a WPT shift NMR library, followed by two optimization steps in producing the predicted molecular composition of a mixture. The robustness of the method was tested on an augmented dataset of 1000 molecular mixtures, each containing 3 to 7 molecules. The method successfully predicted the constituent molecules with a median true positive rate of 1.0 against the varying compositions, while a median false positive rate of 0.04 was obtained. The approach can be scaled easily for much larger datasets.
|
|
|
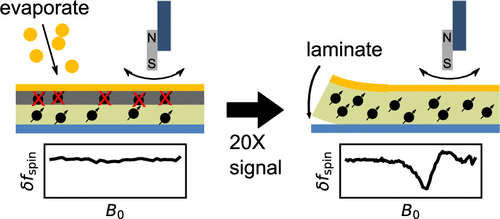
A Non-Perturbative, Low-Noise Surface Coating for Sensitive Force-Gradient Detection of Electron Spin Resonance in Thin Films
M. C. Boucher, C. E. Isaac, P. Sun, P. P. Borbat, and J. A. Marohn
ACS Nano 17, 2c08635 (2023)
Supporting Information
<doi: 10.1021/acsnano.2c08635>
PMID:
36625878
PMCID:
PMC10330945
|
|
|
ABSTRACT: The sensitivity of magnetic resonance force microscopy (MRFM) is limited by surface noise. Coating a thin-film polymer sample with metal has been shown to decrease, by orders of magnitude, sample-related force noise and frequency noise in MRFM experiments. Using both MRFM and inductively detected measurements of electron-spin resonance, we show that thermally evaporating a 12 nm gold layer on a 40 nm nitroxide-doped polystyrene film inactivates the nitroxide spin labels to a depth of 20 nm, making single-spin measurements difficult or impossible. We introduce a "laminated sample" protocol in which the gold layer is first evaporated on a sacrificial polymer. The sample is deposited on the room-temperature gold layer, removed using solvent lift-off, and placed manually on a coplanar waveguide. Electron spin resonance (ESR) of such a laminated sample was detected via MRFM at cryogenic temperatures using a high-compliance cantilever with an integrated 100-nm-scale cobalt tip. A 20-fold increase of spin signal was observed relative to a thin-film sample prepared instead with an evaporated metal coating. The observed signal is still somewhat smaller than expected, and we discuss possible remaining sources of signal loss.
|
|
|
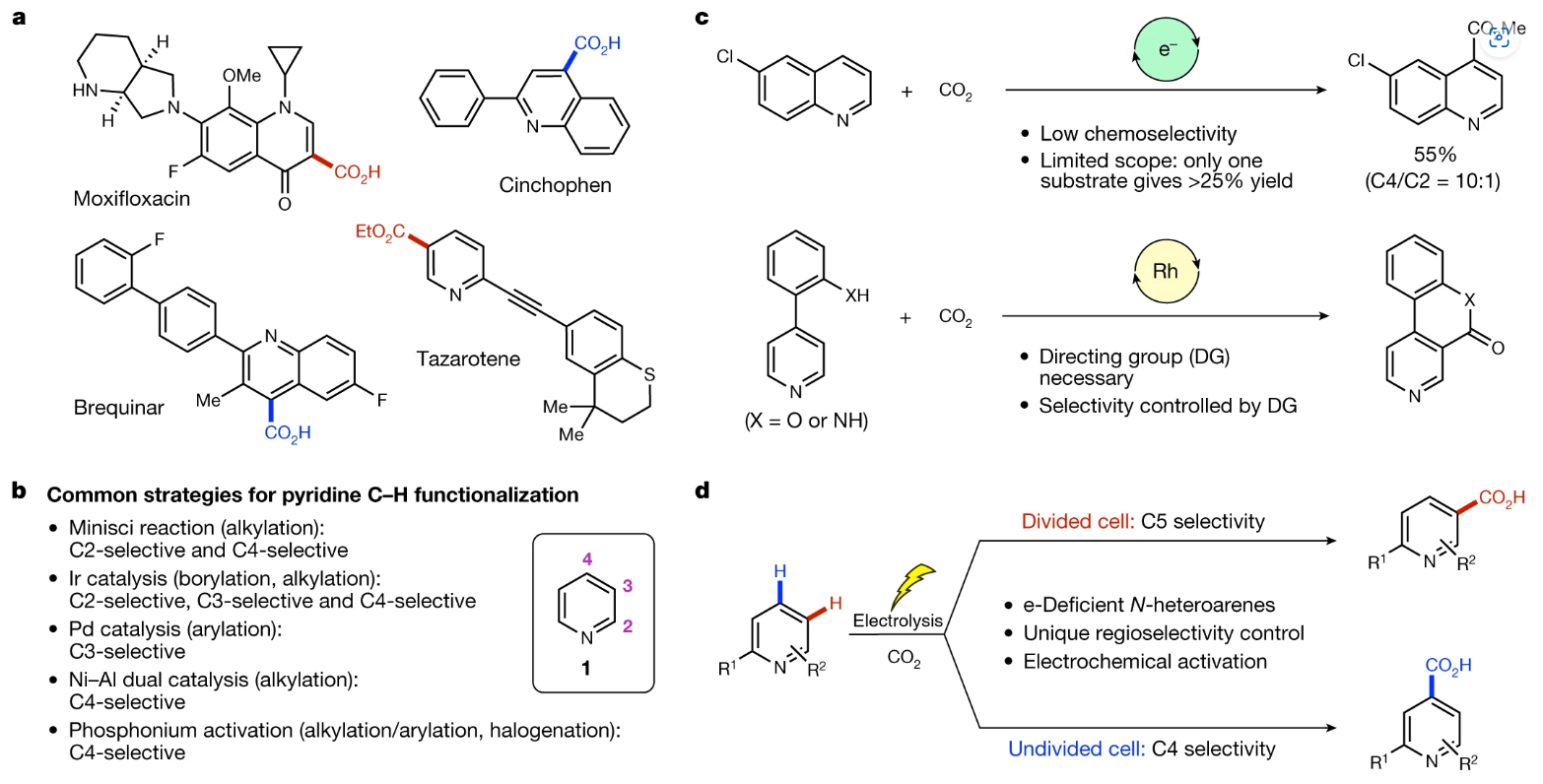
Electrochemical reactor dictates site selectivity in N-heteroarene carboxylations
G.-Q. Sun, P. Yu, W. Zhang, W. Zhang, Y. Wang, L.-L. Liao, Z. Zhang, L. Li, Z. Lu, D.-G. Yu, and S. Lin
Nature 615, 67-72 (2023)
|
|
|
Electrochemical reactor dictates site selectivity in N-heteroarene carboxylations
G.-Q. Sun, P. Yu, W. Zhang, W. Zhang, Y. Wang, L.-L. Liao, Z. Zhang, L. Li, Z. Lu, D.-G. Yu, and S. Lin
Nature 615, 67-72 (2023)
<doi: 10.1038/s41586-022-05667-0>
PMID:
36603811
PMCID:
PMC10036166
|
|
|
ABSTRACT: Pyridines and related N-heteroarenes are commonly found in pharmaceuticals, agrochemicals and other biologically active compounds. Site-selective C–H functionalization would provide a direct way of making these medicinally active products. For example, nicotinic acid derivatives could be made by C–H carboxylation, but this remains an elusive transformation. Here we describe the development of an electrochemical strategy for the direct carboxylation of pyridines using CO2. The choice of the electrolysis setup gives rise to divergent site selectivity: a divided electrochemical cell leads to C5 carboxylation, whereas an undivided cell promotes C4 carboxylation. The undivided-cell reaction is proposed to operate through a paired-electrolysis mechanism, in which both cathodic and anodic events play critical roles in altering the site selectivity. Specifically, anodically generated iodine preferentially reacts with a key radical anion intermediate in the C4-carboxylation pathway through hydrogen-atom transfer, thus diverting the reaction selectivity by means of the Curtin–Hammett principle. The scope of the transformation was expanded to a wide range of N-heteroarenes, including bipyridines and terpyridines, pyrimidines, pyrazines and quinolines.
|
|
|
Analysis of Small-Molecule Mixtures by Super-Resolved 1H NMR Spectroscopy
A. Sinha Roy and M. Srivastava
J. Phys. Chem. A 126, 9108–9113 (2022)
Supporting Information
<doi: 10.1021/acs.jpca.2c06858>
PMID:
36413171
PMCID:
PMC10228708
|

|
|
ABSTRACT: Analysis of small molecules is essential to metabolomics, natural products, drug discovery, food technology, and many other areas of interest. Current barriers preclude from identifying the constituent molecules in a mixture as overlapping clusters of NMR lines pose a major challenge in resolving signature frequencies for individual molecules. While homonuclear decoupling techniques produce much simplified pure shift spectra, they often affect sensitivity. Conversion of typical NMR spectra to pure shift spectra by signal processing without a priori knowledge about the coupling patterns is essential for accurate analysis. We developed a super-resolved wavelet packet transform based 1H NMR spectroscopy that can be used in high-throughput studies to reliably decouple individual constituents of small molecule mixtures. We demonstrate the efficacy of the method on the model mixtures of saccharides and amino acids in the presence of significant noise.
|
|
|
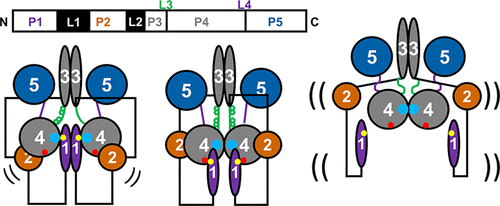
Interdomain Linkers Regulate Histidine Kinase Activity by Controlling Subunit Interactions
Z. Maschmann, S. Chandrasekaran, T. K. Chua, B. R. Crane
Biochemistry 61, 2672-2686 (2022)
Supporting Information
<doi: 10.1021/acs.biochem.2c00326>
PMID:
36321948
PMCID:
PMC10134573
|
|
|
ABSTRACT: Bacterial chemoreceptors regulate the cytosolic multidomain histidine kinase CheA through largely unknown mechanisms. Residue substitutions in the peptide linkers that connect the P4 kinase domain to the P3 dimerization and P5 regulatory domain affect CheA basal activity and activation. To understand the role that these linkers play in CheA activity, the P3-to-P4 linker (L3) and P4-to-P5 linker (L4) were extended and altered in variants of Thermotoga maritima (Tm) CheA. Flexible extensions of the L3 and L4 linkers in CheA-LV1 (linker variant 1) allowed for a well-folded kinase domain that retained wild-type (WT)-like binding affinities for nucleotide and normal interactions with the receptor-coupling protein CheW. However, CheA-LV1 autophosphorylation activity registered ~50-fold lower compared to WT. Neither a WT nor LV1 dimer containing a single P4 domain could autophosphorylate the P1 substrate domain. Autophosphorylation activity was rescued in variants with extended L3 and L4 linkers that favor helical structure and heptad spacing. Autophosphorylation depended on linker spacing and flexibility and not on sequence. Pulse-dipolar electron-spin resonance (ESR) measurements with spin-labeled adenosine 5′-triphosphate (ATP) analogues indicated that CheA autophosphorylation activity inversely correlated with the proximity of the P4 domains within the dimers of the variants. Despite their separation in primary sequence and space, the L3 and L4 linkers also influence the mobility of the P1 substrate domains. In all, interactions of the P4 domains, as modulated by the L3 and L4 linkers, affect domain dynamics and autophosphorylation of CheA, thereby providing potential mechanisms for receptors to regulate the kinase.
|
|
|
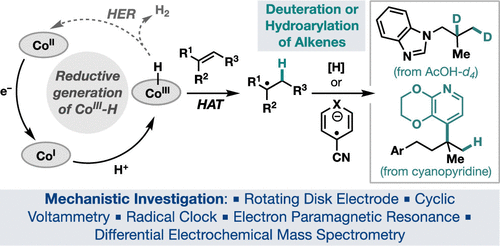
Intercepting Hydrogen Evolution with Hydrogen-Atom Transfer: Electron-Initiated Hydrofunctionalization of Alkenes
X. Wu, C. N. Gannett, J. Liu, R. Zeng, L. F. T. Novaes, H. Wang, H. D. Abruña, S. Lin
J. Am. Chem. Soc. 144, 17783-17791 (2022)
|
|
|
Intercepting Hydrogen Evolution with Hydrogen-Atom Transfer: Electron-Initiated Hydrofunctionalization of Alkenes
X. Wu, C. N. Gannett, J. Liu, R. Zeng, L. F. T. Novaes, H. Wang, H. D. Abruña, S. Lin
J. Am. Chem. Soc. 144, 17783-17791 (2022)
<doi: 10.1021/jacs.2c08278>
PMID:
36137298
|
|
|
ABSTRACT: Hydrogen-atom transfer mediated by earth-abundant transition-metal hydrides (M-Hs) has emerged as a powerful tool in organic synthesis. Current methods to generate M-Hs most frequently rely on oxidatively initiated hydride transfer. Herein, we report a reductive approach to generate Co–H, which allows for canonical hydrogen evolution reactions to be intercepted by hydrogen-atom transfer to an alkene. Electroanalytical and spectroscopic studies provided mechanistic insights into the formation and reactivity of Co–H, which enabled the development of two new alkene hydrofunctionalization reactions.
|
|
|
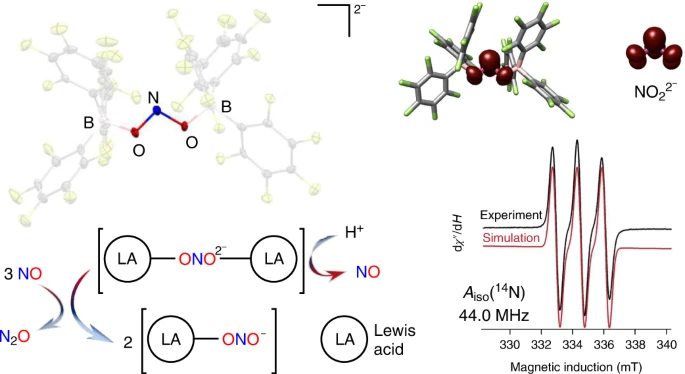
Lewis acid-assisted reduction of nitrite to nitric and nitrous oxides via the elusive nitrite radical dianion
V. Hosseininasab, I. M DiMucci, P. Ghosh, J. A. Bertke, S. Chandrasekharan, C. J. Titus, D. Nordlund, J. H. Freed, K. M. Lancaster, T. H. Warren
Nat. Chem. 14, 1265-1269 (2022)
|
|
|
Lewis acid-assisted reduction of nitrite to nitric and nitrous oxides via the elusive nitrite radical dianion
V. Hosseininasab, I. M DiMucci, P. Ghosh, J. A. Bertke, S. Chandrasekharan, C. J. Titus, D. Nordlund, J. H. Freed, K. M. Lancaster, T. H. Warren
Nat. Chem. 14, 1265-1269 (2022)
Supporting Information
<doi: 10.1038/s41557-022-01025-9>
PMID:
36064970
PMCID:
PMC9633411
|
|
|
ABSTRACT: Reduction of nitrite anions (NO2–) to nitric oxide (NO), nitrous oxide (N2O) and ultimately dinitrogen (N2) takes place in a variety of environments, including in the soil as part of the biogeochemical nitrogen cycle and in acidified nuclear waste. Nitrite reduction typically takes place within the coordination sphere of a redox-active transition metal. Here we show that Lewis acid coordination can substantially modify the reduction potential of this polyoxoanion to allow for its reduction under non-aqueous conditions (–0.74 V versus NHE). Detailed characterization confirms the formation of the borane-capped radical nitrite dianion (NO22–), which features a N(II) oxidation state. Protonation of the nitrite dianion results in the facile loss of nitric oxide (NO), whereas its reaction with NO results in disproportionation to nitrous oxide (N2O) and nitrite (NO2–). This system connects three redox levels in the global nitrogen cycle and provides fundamental insights into the conversion of NO2– to NO.
|
|
|
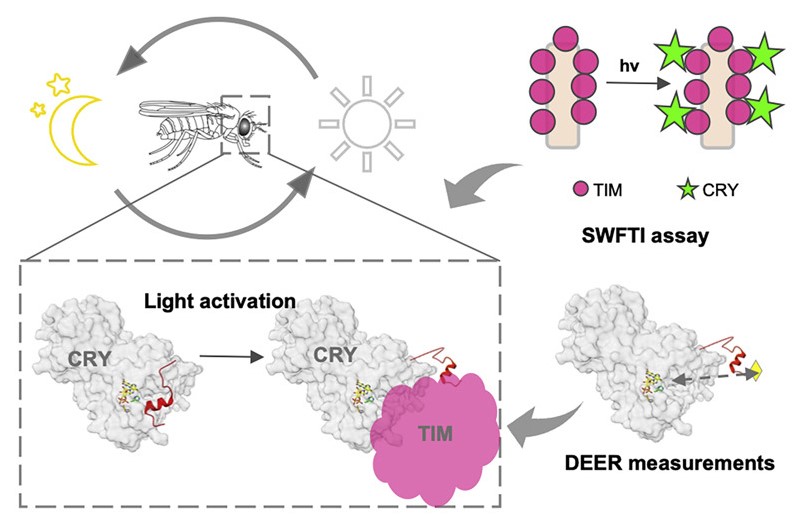
Mechanistic insight into light-dependent recognition of Timeless by Drosophila Cryptochrome
C. Lin, C. M. Schneps, S. Chandrasekaran, A. Ganguly, B. R. Crane
Structure 30, 851-861 (2022)
|
|
|
Mechanistic insight into light-dependent recognition of Timeless by Drosophila Cryptochrome
C. Lin, C. M. Schneps, S. Chandrasekaran, A. Ganguly, B. R. Crane
Structure 30, 851-861 (2022)
<doi: 10.1016/j.str.2022.03.010>
PMID:
35397203
PMCID:
PMC9201872
|
|
|
ABSTRACT: Cryptochrome (CRY) entrains the fly circadian clock by binding to Timeless (TIM) in light. Undocking of a helical C-terminal tail (CTT) in response to photoreduction of the CRY flavin cofactor gates TIM recognition. We present a generally applicable select western-blot-free tagged-protein interaction (SWFTI) assay that allowed the quantification of CRY binding to TIM in dark and light. The assay was used to study CRY variants with residue substitutions in the flavin pocket and correlate their TIM affinities with CTT undocking, as measured by pulse-dipolar ESR spectroscopy and evaluated by molecular dynamics simulations. CRY variants with the CTT removed or undocked bound TIM constitutively, whereas those incapable of photoreduction bound TIM weakly. In response to the flavin redox state, two conserved histidine residues contributed to a robust on/off switch by mediating CTT interactions with the flavin pocket and TIM. Our approach provides an expeditious means to quantify the interactions of difficult-to-produce proteins.
|
|
|
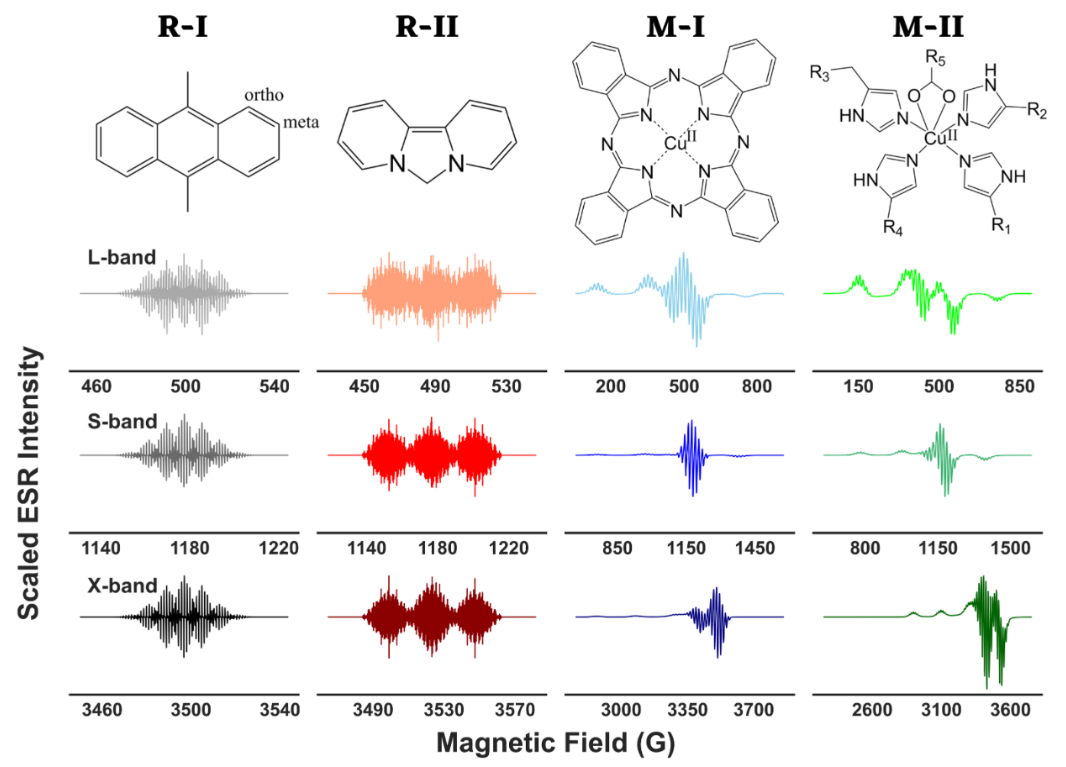
Hyperfine Decoupling of ESR Spectra Using Wavelet Transform
A. Sinha Roy and M. Srivastava
Magnetochemistry 8, 32 (2022)
<doi: 10.3390/magnetochemistry8030032>
PMID:
37475982
PMCID:
PMC10357921
|
|
|
ABSTRACT: The objective of spectral analysis is to resolve and extract relevant features from experimental data in an optimal fashion. In continuous-wave (cw) electron spin resonance (ESR) spectroscopy, both g values of a paramagnetic center and hyperfine splitting (A) caused by its interaction with neighboring magnetic nuclei in a molecule provide important structural and electronic information. However, in the presence of g- and/or A-anisotropy and/or large number of resonance lines, spectral analysis becomes highly challenging. Either high-resolution experimental techniques are employed to resolve the spectra in those cases or a range of suitable ESR frequencies are used in combination with simulations to identify the corresponding g and A values. In this work, we present a wavelet transform technique in resolving both simulated and experimental cw-ESR spectra by separating the hyperfine and super-hyperfine components. We exploit the multiresolution property of wavelet transforms that allow the separation of distinct features of a spectrum based on simultaneous analysis of spectrum and its varying frequency. We retain the wavelet components that stored the hyperfine and/or super-hyperfine features, while eliminating the wavelet components representing the remaining spectrum. We tested the method on simulated cases of metal–ligand adducts at L-, S-, and X-band frequencies, and showed that extracted g values, hyperfine and super-hyperfine coupling constants from simulated spectra, were in excellent agreement with the values of those parameters used in the simulations. For the experimental case of a copper(II) complex with distorted octahedral geometry, the method was able to extract g and hyperfine coupling constant values, and revealed features that were buried in the overlapped spectra.
|
|
|
Highly Basic Clusters in the Herpes Simplex Virus 1 Nuclear Egress Complex Drive Membrane Budding by Inducing Lipid Ordering
M. K. Thorsen, A. Lai, D. P. Hoogerheide, G. C. L. Wong, J. H. Freed, and E. E. Heldwein.
mBio 12 e0154821 (2021)
Supporting Information
<doi: 10.1128/mBio.01548-21>
PMID:
34425706
PMCID:
PMC8406295
|
|
|
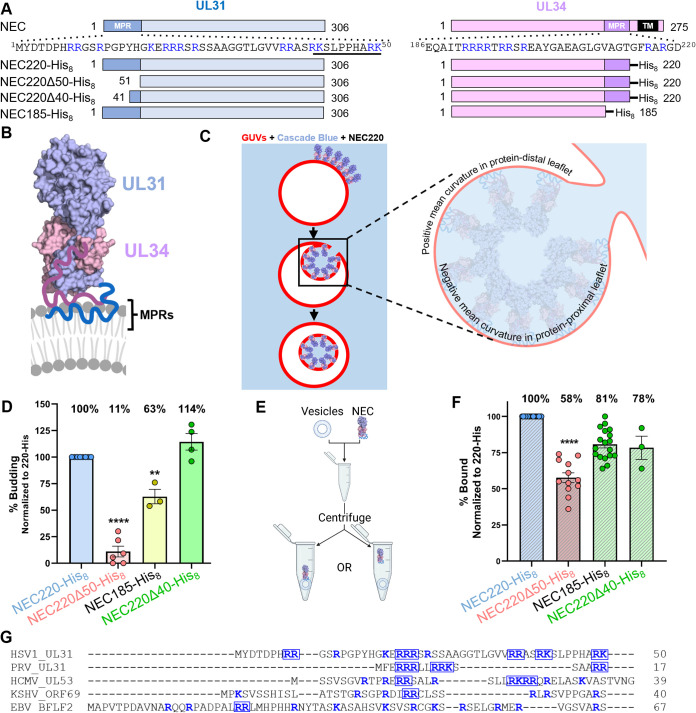
ABSTRACT: During replication of herpesviruses, capsids escape from the nucleus into the cytoplasm by budding at the inner nuclear membrane. This unusual process is mediated by the viral nuclear egress complex (NEC) that deforms the membrane around the capsid by oligomerizing into a hexagonal, membrane‐bound scaffold. Here, we found that highly basic membrane‐proximal regions (MPRs) of the NEC alter lipid order by inserting into the lipid headgroups and promote negative Gaussian curvature. We also find that the electrostatic interactions between the MPRs and the membranes are essential for membrane deformation. One of the MPRs is phosphorylated by a viral kinase during infection, and the corresponding phosphomimicking mutations block capsid nuclear egress. We show that the same phosphomimicking mutations disrupt the NEC‐membrane interactions and inhibit NEC‐mediated budding in vitro, providing a biophysical explanation for the in vivo phenomenon. Our data suggest that the NEC generates negative membrane curvature by both lipid ordering and protein scaffolding and that phosphorylation acts as an off switch that inhibits the membrane‐budding activity of the NEC to prevent capsid‐less budding.
IMPORTANCE Herpesviruses are large viruses that infect nearly all vertebrates and some invertebrates and cause lifelong infections in most of the world's population. During replication, herpesviruses export their capsids from the nucleus into the cytoplasm by an unusual mechanism in which the viral nuclear egress complex (NEC) deforms the nuclear membrane around the capsid. However, how membrane deformation is achieved is unclear. Here, we show that the NEC from herpes simplex virus 1, a prototypical herpesvirus, uses clusters of positive charges to bind membranes and order membrane lipids. Reducing the positive charge or introducing negative charges weakens the membrane deforming ability of the NEC. We propose that the virus employs electrostatics to deform nuclear membrane around the capsid and can control this process by changing the NEC charge through phosphorylation. Blocking NEC-membrane interactions could be exploited as a therapeutic strategy.
|
|
|
SARS-CoV-2 Fusion Peptide has a Greater Membrane Perturbating Effect than SARS-CoV with Highly Specific Dependence on Ca2+
A. L. Lai and J. H. Freed.
J. Mol. Biol. 433, 166946 (2021)
<doi: 10.1016/j.jmb.2021.166946>
PMID:
33744314
PMCID:
PMC7969826
|

|
|
ABSTRACT: Coronaviruses are a major infectious disease threat, and include the zoonotic‐origin human pathogens SARS‐CoV‐2, SARS‐CoV, and MERS‐CoV (SARS‐2, SARS‐1, and MERS). Entry of coronaviruses into host cells is mediated by the spike (S) protein. In our previous ESR studies, the local membrane ordering effect of the fusion peptide (FP) of various viral glycoproteins including the S of SARS‐1 and MERS has been consistently observed. We previously determined that the sequence immediately downstream from the S2′ cleavage site is the bona fide SARS‐1 FP. In this study, we used sequence alignment to identify the SARS‐2 FP, and studied its membrane ordering effect. Although there are only three residue differences, SARS‐2 FP induces even greater membrane ordering than SARS‐1 FP, possibly due to its greater hydrophobicity. This may be a reason that SARS‐2 is better able to infect host cells. In addition, the membrane binding enthalpy for SARS‐2 is greater. Both the membrane ordering of SARS‐2 and SARS‐1 FPs are dependent on Ca2+, but that of SARS‐2 shows a greater response to the presence of Ca2+. Both FPs bind two Ca2+ ions as does SARS‐1 FP, but the two Ca2+ binding sites of SARS‐2 exhibit greater cooperativity. This Ca2+ dependence by the SARS‐2 FP is very ion‐specific. These results show that Ca2+ is an important regulator that interacts with the SARS‐2 FP and thus plays a significant role in SARS‐2 viral entry. This could lead to therapeutic solutions that either target the FP‐calcium interaction or block the Ca2+ channel.
|
|
|
Microsecond dynamics in proteins by two-dimensional ESR. II. Addressing computational challenges
P. Gupta, K. Chaudhari, and J. H. Freed.
J. Chem. Phys. 154, 084115 (2021)
Supporting Information
<doi: 10.1063/5.0042441>
PMID:
33639766
PMCID:
PMC7928224
|
|
|
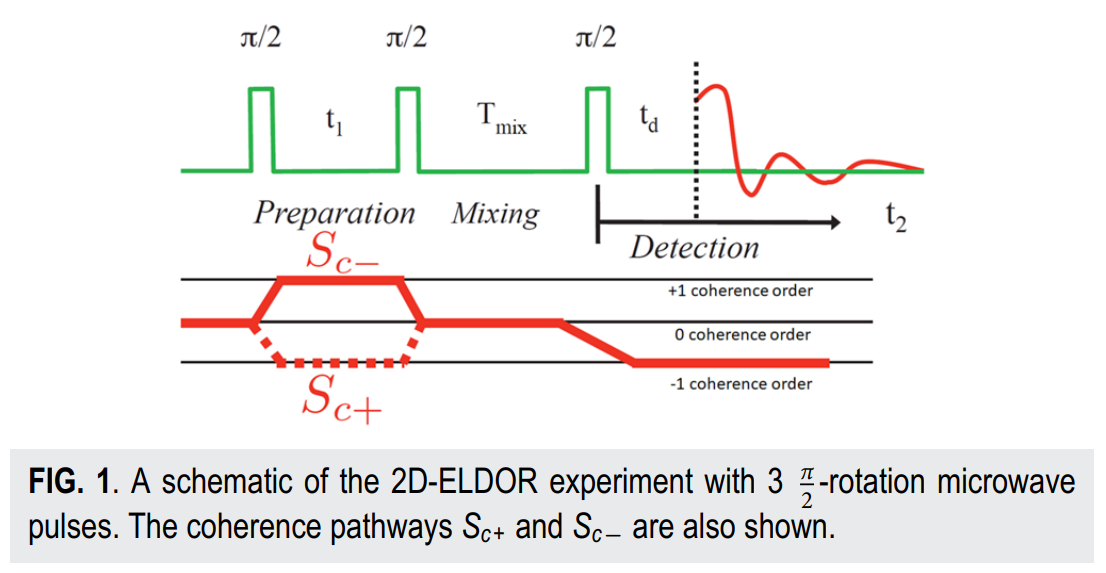
ABSTRACT: Two‐dimensional electron‐electron double resonance (2D‐ELDOR) provides extensive insight into molecular motions. Recent developments permitting experiments at higher frequencies (95 GHz) provide molecular orientational resolution, enabling a clearer description of the nature of the motions. In previous work, we provided simulations for the case of domain motions within proteins that are themselves slowly tumbling in a solution. In order to perform these simulations, it was found that the standard approach of solving the relevant stochastic Liouville equation using the efficient Lanczos algorithm for this case breaks down, so algorithms were employed that rely on the Arnoldi iteration. While they lead to accurate simulations, they are very time‐consuming. In this work, we focus on a variant known as the rational Arnoldi algorithm. We show that this can achieve a significant reduction in computation time. The stochastic Liouville matrix, which is of very large dimension, N, is first reduced to a much smaller dimension, m, e.g., from N ˜ O(104) to m ˜ 60, that spans the relevant Krylov subspace from which the spectrum is predicted. This requires the selection of the m frequency shifts to be utilized. A method of adaptive shift choice is introduced to optimize this selection. We also find that these procedures help in optimizing the pruning procedure that greatly reduces the dimension of the initial N dimensional stochastic Liouville matrix in such subsequent computations.
|
|
|
Microsecond Exchange Processes Studied by Two‐Dimensional ESR at 95 GHz
B. Dzikovski, V. V. Khramtsov, S. Chandrasekaran, C. Dunnam, M. Shah, and J. H. Freed.
J. Am. Chem. Soc. 142, 21368-21381 (2020)
Supporting Information
<doi: 10.1021/jacs.0c09469>
PMID:
33305945
PMCID:
PMC7810061
|
|
|
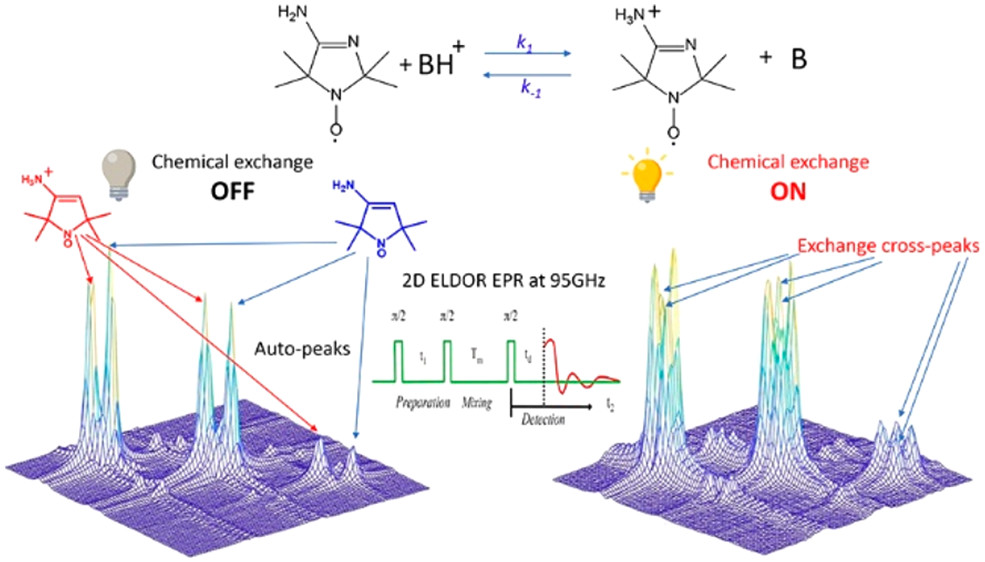
ABSTRACT: Exchange processes which include conformational change, protonation/deprotonation, and binding equilibria are routinely studied by 2D exchange NMR techniques, where information about the exchange of nuclei between environments with different NMR shifts is obtained from the development of cross‐peaks. Whereas 2D NMR enables the real time study of millisecond and slower exchange processes, 2D ESR in the form of 2D‐ELDOR (two‐dimensional electron‐electron double resonance) has the potential for such studies over the nanosecond to microsecond real time scales. Cross‐peak development due to chemical exchange has been seen previously for semiquinones in ESR, but this is not possible for most common ESR probes, such as nitroxides, studied at typical ESR frequencies because, unlike NMR, the exchanging states yield ESR signals that are not resolved from each other within their respective line widths. But at 95 GHz, it becomes possible to resolve them in many cases because of the increased g‐factor resolution. The 95 GHz instrumental developments occurring at ACERT now enable such studies. We demonstrate these new capabilities in two studies: (A) the protonation/deprotonation process for a pH‐sensitive imidazoline spin label in aqueous solution where the exchange rate and the population ratio of the exchanging states are controlled by the concentration and pH of the buffer solution, respectively, and (B) a nitroxide radical partitioning between polar (aqueous) and nonpolar (phospholipid) environments in multilamellar lipid vesicles, where the cross‐peak development arises from the exchange of the nitroxide between the two phases. This work represents the first example of the observation and analysis of cross‐peaks arising from chemical exchange processes involving nitroxide spin labels.
|
|
|
Engineered chemotaxis core signaling units indicate a constrained kinase-off state
A. R. Muok, T. K. Chua, M. Srivastava, W. Yang, Z. Maschmann, P. P. Borbat, J. Chong, S. Zhang, J. H. Freed, A. Briegel, B. R. Crane.
Sci. Signal. 13, eabc1328 (2020)
|
|
|
Engineered chemotaxis core signaling units indicate a constrained kinase-off state
A. R. Muok, T. K. Chua, M. Srivastava, W. Yang, Z. Maschmann, P. P. Borbat, J. Chong, S. Zhang, J. H. Freed, A. Briegel, B. R. Crane.
Sci. Signal. 13, eabc1328 (2020)
Supporting Information
<doi: 10.1126/scisignal.abc1328>
PMID:
33172954
PMCID:
PMC7790435
|
|
|
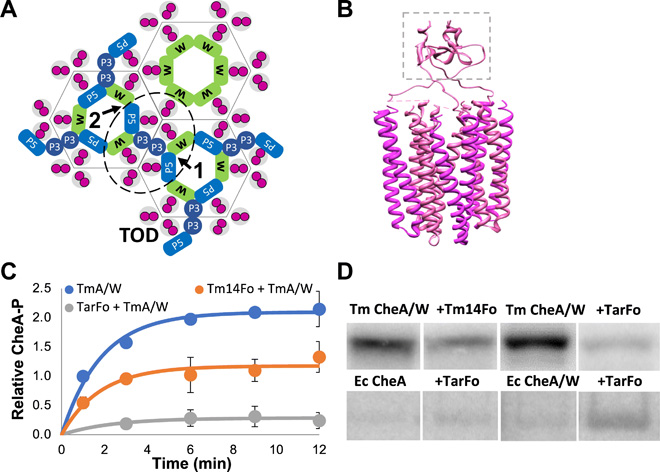
ABSTRACT: Bacterial chemoreceptors, the histidine kinase CheA, and the coupling protein CheW form transmembrane molecular arrays with remarkable sensing properties. The receptors inhibit or stimulate CheA kinase activity depending on the presence of attractants or repellants, respectively. We engineered chemoreceptor cytoplasmic regions to assume a trimer of receptor dimers configuration that formed well‐defined complexes with CheA and CheW and promoted a CheA kinase‐off state. These mimics of core signaling units were assembled to homogeneity and investigated by site‐directed spin‐labeling with pulse‐dipolar electron‐spin resonance spectroscopy (PDS), small‐angle x‐ray scattering, targeted protein cross‐linking, and cryo–electron microscopy. The kinase‐off state was especially stable, had relatively low domain mobility, and associated the histidine substrate and docking domains with the kinase core, thus preventing catalytic activity. Together, these data provide an experimentally restrained model for the inhibited state of the core signaling unit and suggest that chemoreceptors indirectly sequester the kinase and substrate domains to limit histidine autophosphorylation.
|
|
|
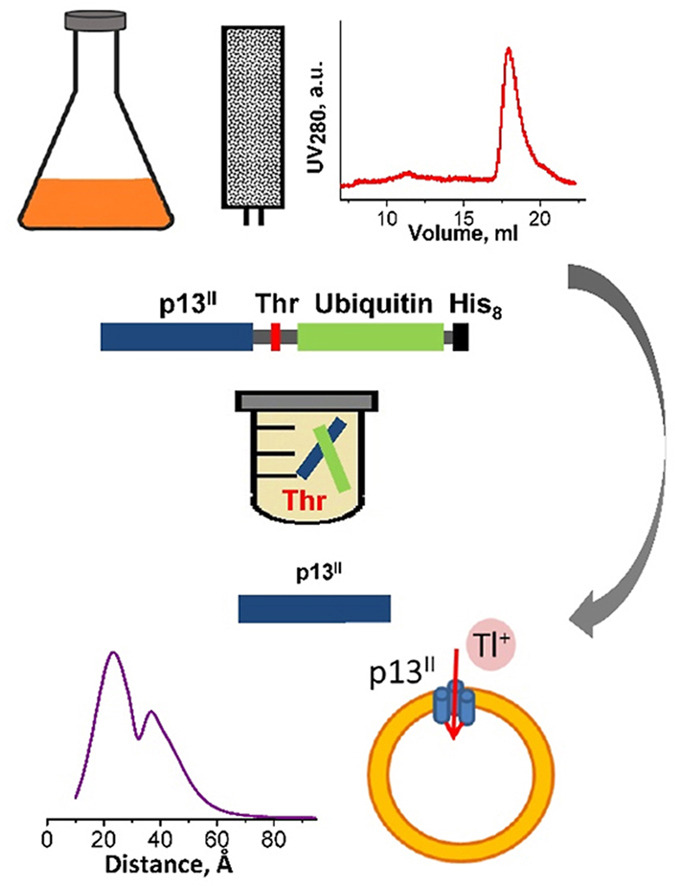
High-yield production in E. coli and characterization of full-length functional p13II protein from human T-cell leukemia virus type 1
E. R. Georgieva, P. P. Borbat, C. Fanouraki, J. H. Freed.
Protein Expr. Purif. 173, 105659 (2020)
Supporting Information
<doi: 10.1016/j.pep.2020.105659>
PMID:
32360379
PMCID:
PMC7266171
|
|
|
ABSTRACT: Human T-cell leukemia virus type 1 is an oncovirus that causes aggressive adult T-cell leukemia but is also responsible for severe neurodegenerative and endocrine disorders. Combatting HTLV-1 infections requires a detailed understanding of the viral mechanisms in the host. Therefore, in vitro studies of important virus-encoded proteins would be critical. Our focus herein is on the HTLV-1-encoded regulatory protein p13II, which interacts with the inner mitochondrial membrane, increasing its permeability to cations (predominantly potassium, K+). Thereby, this protein affects mitochondrial homeostasis. We report on our progress in developing specific protocols for heterologous expression of p13II in E. coli, and methods for its purification and characterization. We succeeded in producing large quantities of highly-pure full-length p13II, deemed to be its fully functional form. Importantly, our particular approach based on the fusion of ubiquitin to the p13II C-terminus was instrumental in increasing the persistently low expression of soluble p13II in its native form. We subsequently developed approaches for protein spin labeling and a conformation study using double electron-electron resonance (DEER) spectroscopy and a fluorescence-based cation uptake assay for p13II in liposomes. Our DEER results point to large protein conformation changes occurring upon transition from the soluble to the membrane-bound state. The functional assay on p13II-assisted transport of thallium (Tl+) through the membrane, wherein Tl+ substituted for K+, suggests transmembrane potential involvement in p13II function. Our study lays the foundation for expansion of in vitro functional and structural investigations on p13II and would aid in the development of structure-based protein inhibitors and markers.
|
|
|
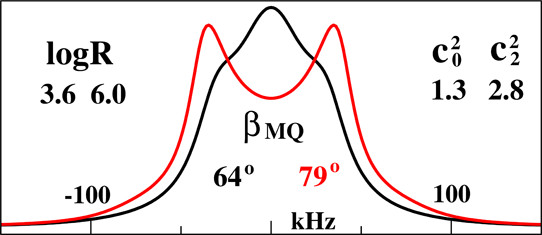
Structural Dynamics by NMR in the Solid State: The Unified MOMD Perspective Applied to Organic Frameworks with Interlocked Molecules
E. Meirovitch, Z. Liang, and J. H. Freed.
J. Phys. Chem. B 124, 6225-6235 (2020)
Supporting Information
<doi: 10.1021/acs.jpcb.0c03687>
PMID:
32584038
PMCID:
PMC7666760
|
|
|
ABSTRACT: The microscopic-order-macroscopic-disorder (MOMD) approach for NMR lineshape analysis has been applied to the University of Windsor Dynamic Materials (UWDM) of types 1, 2, α-3, β-3, and 5, which are metal–organic frameworks (MOFs) comprising mobile mechanically interlocked molecules (MIMs). The mobile MIM components are selectively deuterated crown ether macrocycles – 24C6, 22C6, and B24C6. Their motion is described in MOMD by an effective/collective dynamic mode characterized by a diffusion tensor, R, a restricting/ordering potential, u, expanded in the Wigner rotation matrix elements, D0,KL, and features of local geometry. Experimental 2H lineshapes are available over 220 K (on average) and in some cases 320 K. They are reproduced with axial R, u given by the terms D0,02 and D0,|2|2, and established local geometry. For UWDM of types 1, β-3, and 5, where the macrocycle resides in a relatively loose space, u is in the 1–3 kT, R∥ in the (1.0–2.5) × 106 s–1, and R⊥ in the (0.4–2.5) × 104 s–1 range; the deuterium atom is bonded to a carbon atom with tetrahedral coordination character. For UWDM of types 2 and α-3, where the macrocycle resides in a much tighter space, a substantial change in the symmetry of u and the coordination character of the 2H-bonded carbon are detected at higher temperatures. The activation energies for R∥ and R⊥ are characteristic of each system. The MOMD model is general; effective/collective dynamic modes are treated. The characteristics of motion, ordering, and geometry are physically well-defined; they differ from case to case in extent and symmetry but not in essence. Physical clarity and consistency provide new insights. A previous interpretation of the same experimental data used models consisting of collections of independent simple motions. These models are specific to each case and temperature. Within their scope, generating consistent physical pictures and comparing cases are difficult; possible collective modes are neglected.
|
|
|
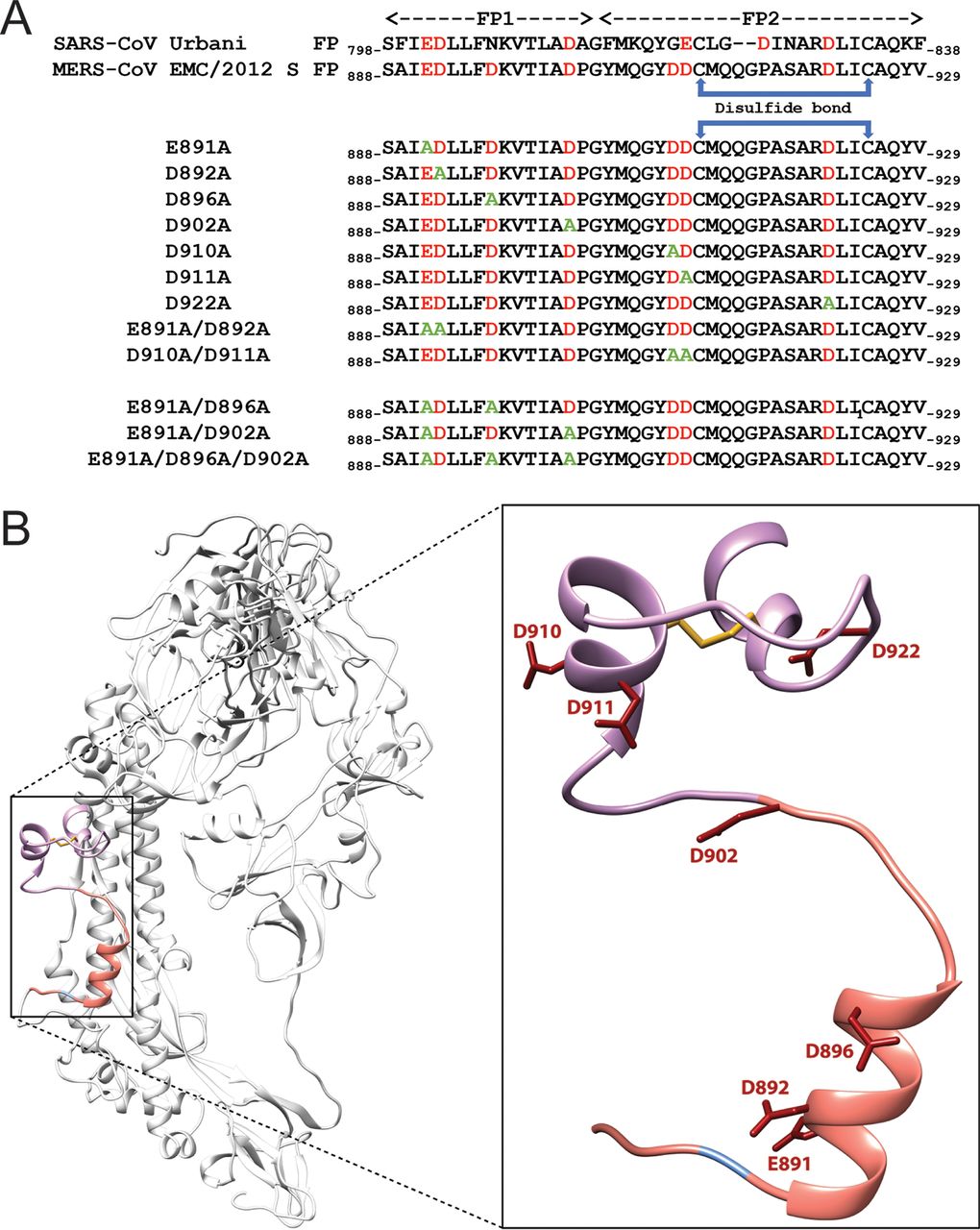
Ca2+ Ions Promote Fusion of Middle East Respiratory Syndrome Coronavirus with Host Cells and Increase Infectivity
M. R. Straus, T. Tang, A. L. Lai, A. Flegel, M. Bidon, J. H. Freed, S. Daniel, G. R. Whittaker.
J. Virol. 94, e00426-20 (2020)
Supporting Information
<doi: 10.1128/JVI.00426-20>
PMID:
32295925
PMCID:
PMC7307142
|
|
|
ABSTRACT: Fusion with, and subsequent entry into, the host cell is one of the critical steps in the life cycle of enveloped viruses. For Middle East respiratory syndrome coronavirus (MERS-CoV), the spike (S) protein is the main determinant of viral entry. Proteolytic cleavage of the S protein exposes its fusion peptide (FP), which initiates the process of membrane fusion. Previous studies on the related severe acute respiratory syndrome coronavirus (SARS-CoV) FP have shown that calcium ions (Ca2+) play an important role in fusogenic activity via a Ca2+ binding pocket with conserved glutamic acid (E) and aspartic acid (D) residues. SARS-CoV and MERS-CoV FPs share a high sequence homology, and here, we investigated whether Ca2+ is required for MERS-CoV fusion by screening a mutant array in which E and D residues in the MERS-CoV FP were substituted with neutrally charged alanines (A). Upon verifying mutant cell surface expression and proteolytic cleavage, we tested their ability to mediate pseudoparticle (PP) infection of host cells in modulating Ca2+ environments. Our results demonstrate that intracellular Ca2+ enhances MERS-CoV wild-type (WT) PP infection by approximately 2-fold and that E891 is a crucial residue for Ca2+ interaction. Subsequent electron spin resonance (ESR) experiments revealed that this enhancement could be attributed to Ca2+ increasing MERS-CoV FP fusion-relevant membrane ordering. Intriguingly, isothermal calorimetry showed an approximate 1:1 MERS-CoV FP to Ca2+ ratio, as opposed to an 1:2 SARS-CoV FP to Ca2+ ratio, suggesting significant differences in FP Ca2+ interactions of MERS-CoV and SARS-CoV FP despite their high sequence similarity.
IMPORTANCE Middle East respiratory syndrome coronavirus (MERS-CoV) is a major emerging infectious disease with zoonotic potential and has reservoirs in dromedary camels and bats. Since its first outbreak in 2012, the virus has repeatedly transmitted from camels to humans, with 2,468 confirmed cases causing 851 deaths. To date, there are no efficacious drugs and vaccines against MERS-CoV, increasing its potential to cause a public health emergency. In order to develop novel drugs and vaccines, it is important to understand the molecular mechanisms that enable the virus to infect host cells. Our data have found that calcium is an important regulator of viral fusion by interacting with negatively charged residues in the MERS-CoV FP region. This information can guide therapeutic solutions to block this calcium interaction and also repurpose already approved drugs for this use for a fast response to MERS-CoV outbreaks.
|
|
|
Microsecond dynamics in proteins by two-dimensional ESR: Predictions
P. Gupta, Z. Liang, and J. H. Freed.
J. Chem. Phys. 152, 214112 (2020)
Supporting Information
<doi: 10.1063/5.0008094>
PMID:
32505151
PMCID:
PMC7863697
|

|
|
ABSTRACT: Two-dimensional electron-electron double resonance (2D-ELDOR) provides extensive insight into molecular motions. Recent developments permitting experiments at higher frequencies (95 GHz) provide molecular orientational resolution, enabling a clearer description of the nature of the motions. In this work, simulations are provided for the example of domain motions within proteins that are themselves slowly tumbling in solution. These show the nature of the exchange cross-peaks that are predicted to develop in real time from such domain motions. However, we find that the existing theoretical methods for computing 2D-ELDOR experiments over a wide motional range begin to fail seriously when applied to very slow motions characteristic of proteins in solution. One reason is the failure to obtain accurate eigenvectors and eigenvalues of the complex symmetric stochastic Liouville matrices describing the experiment when computed by the efficient Lanczos algorithm in the range of very slow motion. Another, perhaps more serious, issue is that these matrices are "non-normal," such that for the very slow motional range even rigorous diagonalization algorithms do not yield the correct eigenvalues and eigenvectors. We have employed algorithms that overcome both these issues and lead to valid 2D-ELDOR predictions even for motions approaching the rigid limit. They are utilized to describe the development of cross-peaks in 2D-ELDOR at 95 GHz for a particular case of domain motion.
|
|
|
Conformational Dynamics in Extended RGD-Containing Peptides
W. R. Lindemann, A. J. Mijalis, J. L. Alonso, P. P. Borbat, J. H. Freed, M. A. Arnaout, B. L. Pentelute, and J. H. Ortony.
Biomacromolecules 21, 2786-2794 (2020)
Supporting Information
<doi: 10.1021/acs.biomac.0c00506>
PMID:
32469507
PMCID:
PMC7388056
|
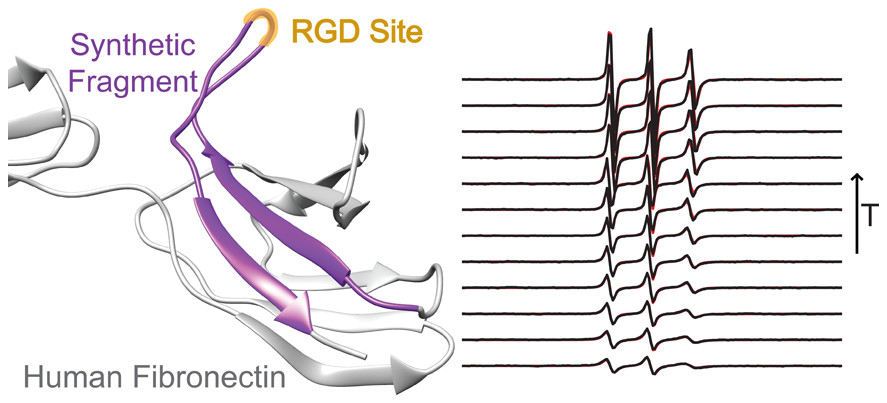
|
|
ABSTRACT: RGD is a prolific example of a tripeptide used in biomaterials for cell adhesion, but the potency of free or surface-bound RGD tripeptide is orders-of-magnitude less than the RGD domain within natural proteins. We designed a set of peptides with varying lengths, composed of fragments of fibronectin protein whose central three residues are RGD, in order to vary their conformational behavior without changing the binding site's chemical environment. With these peptides, we measure the conformational dynamics and transient structure of the active site. Our studies reveal how flanking residues affect conformational behavior and integrin binding. We find that disorder of the binding site is important to the potency of RGD peptides and that transient hydrogen bonding near the RGD site affects both the energy landscape roughness of the peptides and peptide binding. This phenomenon is independent of longer-range folding interactions and helps explain why short binding sequences, including RGD itself, do not fully replicate the integrin-targeting properties of extracellular matrix proteins. Our studies reinforce that peptide binding is a holistic event and fragments larger than those directly involved in binding should be considered in the design of peptide epitopes for functional biomaterials.
|
|
|
George K. Fraenkel, Electron Spin Resonance Pioneer
J.H. Freed.
In Pioneers of Magnetic Resonance. Strom, E. T., Mainz, V. V., Eds. American Chemical Society: Washington, DC, ACS Symposium Series, 2020; Volume 1349, Chapter 8, pp. 137-154.
|
|
|
George K. Fraenkel, Electron Spin Resonance Pioneer
J.H. Freed.
In Pioneers of Magnetic Resonance. Strom, E. T., Mainz, V. V., Eds. American Chemical Society: Washington, DC, ACS Symposium Series, 2020; Volume 1349, Chapter 8, pp. 137-154.
<doi: 10.1021/bk-2020-1349.ch008>
PMID: [None-book] PMCID:
[None-book]
|
|
|
ABSTRACT: George K. Fraenkel (1921-2009), although less known today, was one of the leading pioneers in the development and use of Electron Spin Resonance (ESR) techniques for studying the structure and dynamical interactions of molecules. Together with his students he developed high-sensitivity, high-resolution spectrometers that enabled them to pioneer the study of ESR in free radicals in solution. His work provided breakthroughs that led to advances in several fields of chemistry, and laid the foundations for later research on the properties of biological systems. This chapter provides a thematic overview, arranged chronologically, of his various studies. Short descriptions are provided of his achievements in these areas, based on his most important papers, so that present-day researchers can appreciate the extent of his accomplishments.
|
|
|
Calcium Ions Directly Interact with the Ebola Virus Fusion Peptide To Promote Structure–Function Changes That Enhance Infection
L. Nathan, A. L. Lai, J. K. Millet, M. R. Straus, J. H. Freed, G. R. Whittaker, and S. Daniel.
ACS Infect. Dis. 6, 250-260 (2020)
Supporting Information
<doi: 10.1021/acsinfecdis.9b00296>
PMID:
31746195
PMCID:
PMC7040957
|
|
|
ABSTRACT: Ebola virus disease is a serious global health concern given its periodic occurrence, high lethality, and the lack of approved therapeutics. Certain drugs that alter intracellular calcium, particularly in endolysosomes, have been shown to inhibit Ebola virus infection; however, the underlying mechanism is unknown. Here, we provide evidence that Zaire ebolavirus (EBOV) infection is promoted in the presence of calcium as a result of the direct interaction of calcium with the EBOV fusion peptide (FP). We identify the glycoprotein residues D522 and E540 in the FP as functionally critical to EBOV's interaction with calcium. We show using spectroscopic and biophysical assays that interactions of the fusion peptide with Ca2+directly targets the Ebola virus fusion peptide and influences its conformation. As these residues are highly conserved across the Filoviridae, calcium's impact on fusion, and subsequently infectivity, is a key interaction that can be leveraged for developing strategies to defend against Ebola infection. This mechanistic insight provides a rationale for the use of calcium-interfering drugs already approved by the FDA as therapeutics against Ebola and enables further development of novel drugs to combat the virus.
|
|
|
The asymmetric function of Dph1–Dph2 heterodimer in diphthamide biosynthesis
M. Dong, E. E. Dando, I. Kotliar, X. Su, B. Dzikovski, J. H. Freed, and H. Lin.
J. Biol. Inorg. Chem. 24, 777-782 (2019)
<doi: 10.1007/s00775-019-01702-0>
PMID:
31463593
PMCID:
PMC6893874
|

|
|
ABSTRACT: Diphthamide, the target of diphtheria toxin, is a post-translationally modified histidine residue found in archaeal and eukaryotic translation elongation factor 2 (EF2). In the first step of diphthamide biosynthesis, a [4Fe–4S] cluster-containing radical SAM enzyme, Dph1–Dph2 heterodimer in eukaryotes or Dph2 homodimer in archaea, cleaves S-adenosylmethionine and transfers the 3-amino-3-carboxypropyl group to EF2. It was demonstrated previously that for the archaeal Dph2 homodimer, only one [4Fe–4S] cluster is necessary for the in vitro activity. Here, we demonstrate that for the eukaryotic Dph1–Dph2 heterodimer, the [4Fe–4S] cluster-binding cysteine residues in each subunit are required for diphthamide biosynthesis to occur in vivo. Furthermore, our in vitro reconstitution experiments with Dph1–Dph2 mutants suggested that the Dph1 cluster serves a catalytic role, while the Dph2 cluster facilitates the reduction of the Dph1 cluster by the physiological reducing system Dph3/Cbr1/NADH. Our results reveal the asymmetric functional roles of the Dph1–Dph2 heterodimer and may help to understand how the Fe–S clusters in radical SAM enzymes are reduced in biology.
|
|
|
Local ordering and dynamics in anisotropic media by magnetic resonance: from liquid crystals to proteins
E. Meirovitch & J. H. Freed.
Liq. Cryst. 47, 1926-1954 (2020)
<doi: 10.1080/02678292.2019.1622158>
PMID:
32435078
PMCID:
PMC7239324
|
|
|
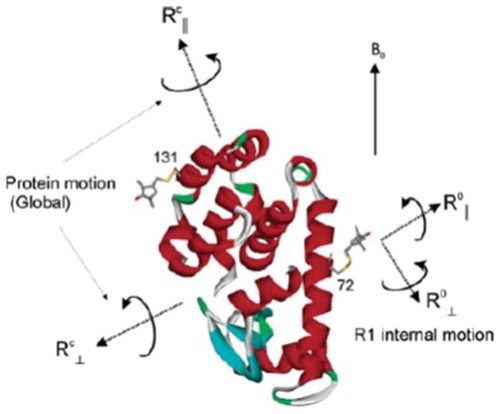
ABSTRACT: Magnetic resonance methods have been used extensively for over 50 years to elucidate molecular structure and dynamics of liquid crystals (LCs), providing information quite unique in its rigour and extent. The ESR- or NMR-active probe is often a solute molecule reporting on characteristics associated with the surrounding (LC) medium, which exerts the spatial restrictions on the probe. The theoretical approaches developed for LCs are applicable to anisotropic media in general. Of particular interest is the interior space of a globular protein labelled, e.g. with a nitroxide moiety or a 15N–1H bond. The ESR or NMR label plays the role of the probe and the internal protein surroundings the role of the anisotropic medium. A general feature of the restricted motions is the local ordering, i.e. the nature, magnitude and symmetry of the spatial restraints exerted at the site of the moving probe. This property is the main theme of the present review article. We outline its treatment in our work from both the theoretical and the experimental points of view, highlighting the new physical insights gained. Our illustrations include studies on thermotropic (nematic and smectic) and lyotropic liquid crystals formed by phospholipids, in addition to studies of proteins.
|
|
|
Insights into histidine kinase activation mechanisms from the monomeric blue light sensor EL346
I. Dikiy, U. R. Edupuganti, R. R. Abzalimov, P. P. Borbat, M. Srivastava, J. H. Freed, and K. H. Gardner.
Proc. Natl. Acad. Sci. 116, 4963-4972 (2019)
Supporting Information
<doi: 10.1073/pnas.1813586116>
PMID:
30808807
PMCID:
PMC6421462
|

|
|
SIGNIFICANCE: All living things must sense and react to their environment. Many single-celled organisms do so by using two-component systems, most simply consisting of a sensor histidine kinase and a response regulator. These systems are involved in pathogenicity pathways and can be targeted by new antibiotics. However, the molecular mechanisms used by histidine kinases to translate sensing into responses are not well understood. To probe this general question, we apply a combination of biophysical techniques to a monomeric histidine kinase that senses blue light to determine the structural changes occurring upon activation. We find these changes to be similar to those predicted for the common dimeric histidine kinases, illustrating that the mechanism of activation is conserved regardless of oligomeric state.
ABSTRACT: Translation of environmental cues into cellular behavior is a necessary process in all forms of life. In bacteria, this process frequently involves two-component systems in which a sensor histidine kinase (HK) autophosphorylates in response to a stimulus before subsequently transferring the phosphoryl group to a response regulator that controls downstream effectors. Many details of the molecular mechanisms of HK activation are still unclear due to complications associated with the multiple signaling states of these large, multidomain proteins. To address these challenges, we combined complementary solution biophysical approaches to examine the conformational changes upon activation of a minimal, blue-light–sensing histidine kinase from Erythrobacter litoralis HTCC2594, EL346. Our data show that multiple conformations coexist in the dark state of EL346 in solution, which may explain the enzyme's residual dark-state activity. We also observe that activation involves destabilization of the helices in the dimerization and histidine phosphotransfer-like domain, where the phosphoacceptor histidine resides, and their interactions with the catalytic domain. Similar light-induced changes occur to some extent even in constitutively active or inactive mutants, showing that light sensing can be decoupled from activation of kinase activity. These structural changes mirror those inferred by comparing X-ray crystal structures of inactive and active HK fragments, suggesting that they are at the core of conformational changes leading to HK activation. More broadly, our findings uncover surprising complexity in this simple system and allow us to outline a mechanism of the multiple steps of HK activation.
|
|
|
|
|
Cofactors are essential constituents of stable and seeding-active tau fibrils
Y. Fichou, Y. Lin, J. N. Rauch, M. Vigers, Z. Zeng, M. Srivastava, T. J. Keller, J. H. Freed, K. S. Kosik, and S. Han.
Proc. Natl. Acad. Sci. 115, 13234-13239 (2018)
|

|
|
Cofactors are essential constituents of stable and seeding-active tau fibrils
Y. Fichou, Y. Lin, J. N. Rauch, M. Vigers, Z. Zeng, M. Srivastava, T. J. Keller, J. H. Freed, K. S. Kosik, and S. Han.
Proc. Natl. Acad. Sci. 115, 13234-13239 (2018)
Supporting Information
<doi: 10.1073/pnas.1810058115>
PMID:
30538196
PMCID:
PMC6310788
|
|
|
SIGNIFICANCE: The tau protein is involved in Alzheimer's and other neurodegenerative diseases, where the location, morphology, and quantity of amyloid fibrils composed of tau correlate with the disease type and stage. While tau fibrillary aggregates have been colocalized in brains together with several cofactors, their role in fibril formation, structure, and seeding has been largely neglected. We show that seeding of tau aggregation is facilitated by polyanionic cofactors, and that seeded or recombinant mature fibrils depolymerize into monomers when their cofactor is removed. We show that cofactor-assisted seeding with mouse brain-derived tau fibrils yielded tau fibrils with distinct and narrowed structural properties compared with heparin-induced fibrils, suggesting that the fibrillar templates tuned the structure of the seeded fibril.
ABSTRACT: Amyloid fibrils are cross-β–rich aggregates that are exceptionally stable forms of protein assembly. Accumulation of tau amyloid fibrils is involved in many neurodegenerative diseases, including Alzheimer's disease (AD). Heparin-induced aggregates have been widely used and assumed to be a good tau amyloid fibril model for most biophysical studies. Here we show that mature fibrils made of 4R tau variants, prepared with heparin or RNA, spontaneously depolymerize and release monomers when their cofactors are removed. We demonstrate that the cross-β-sheet assembly formed in vitro with polyanion addition is unstable at room temperature. We furthermore demonstrate high seeding capacity with transgenic AD mouse brain-extracted tau fibrils in vitro that, however, is exhausted after one generation, while supplementation with RNA cofactors resulted in sustained seeding over multiple generations. We suggest that tau fibrils formed in brains are supported by unknown cofactors and inhere higher-quality packing, as reflected in a more distinct conformational arrangement in the mouse fibril-seeded, compared with heparin-induced, tau fibrils. Our study suggests that the role of cofactors in tauopathies is a worthy focus of future studies, as they may be viable targets for diagnosis and therapeutics.
|
|
|
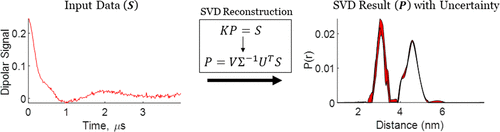
Singular Value Decomposition Method To Determine Distance Distributions in Pulsed Dipolar Electron Spin Resonance: II. Estimating Uncertainty
M. Srivastava and J. H. Freed.
J. Phys. Chem. A 123, 359-370 (2019)
Supporting Information
<doi: 10.1021/acs.jpca.8b07673>
PMID:
30525624
PMCID:
PMC6372347
|
|
|
ABSTRACT: This paper is a continuation of the method introduced by Srivastava and Freed (2017) that is a new method based on truncated singular value decomposition (TSVD) for obtaining physical results from experimental signals without any need for Tikhonov regularization or other similar methods that require a regularization parameter. We show here how to estimate the uncertainty in the SVD-generated solutions. The uncertainty in the solution may be obtained by finding the minimum and maximum values over which the solution remains converged. These are obtained from the optimum range of singular value contributions, where the width of this region depends on the solution point location (e.g., distance) and the signal-to-noise ratio (SNR) of the signal. The uncertainty levels typically found are very small with substantial SNR of the (denoised) signal, emphasizing the reliability of the method. With poorer SNR, the method is still satisfactory but with greater uncertainty, as expected. Pulsed dipolar electron spin resonance spectroscopy experiments are used as an example, but this TSVD approach is general and thus applicable to any similar experimental method wherein singular matrix inversion is needed to obtain the physically relevant result. We show that the Srivastava–Freed TSVD method along with the estimate of uncertainty can be effectively applied to pulsed dipolar electron spin resonance signals with SNR > 30, and even for a weak signal (e.g., SNR ≈ 3) reliable results are obtained by this method, provided the signal is first denoised using wavelet transforms (WavPDS).
|
|
|
Site-Specific Incorporation of a Cu2+ Spin Label into Proteins for Measuring Distances by Pulsed Dipolar Electron Spin Resonance Spectroscopy
G. E. Merz, P. P. Borbat, A. R. Muok, M. Srivastava, D. N. Bunck, J. H. Freed, and B. R. Crane.
J. Phys. Chem. B 122, 9443-9451 (2018)
Supporting Information
<doi: 10.1021/acs.jpcb.8b05619>
PMID:
30222354
PMCID:
PMC6215709
|
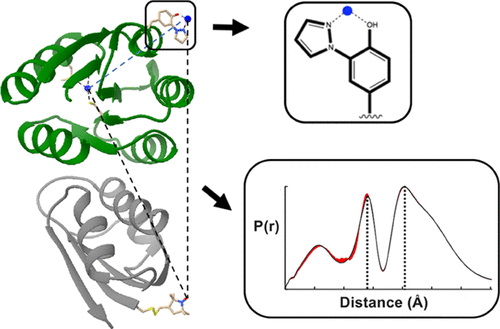
|
|
ABSTRACT: Pulsed dipolar electron spin resonance spectroscopy (PDS) is a powerful tool for measuring distances in solution-state macromolecules. Paramagnetic metal ions, such as Cu2+, are used as spin probes because they can report on metalloprotein features and can be spectroscopically distinguished from traditional nitroxide (NO)-based labels. Here, we demonstrate site-specific incorporation of Cu2+ into non-metalloproteins through the use of a genetically encodable non-natural amino acid, 3-pyrazolyltyrosine (PyTyr). We first incorporate PyTyr in cyan fluorescent protein to measure Cu2+-to-NO distances and examine the effects of solvent conditions on Cu2+ binding and protein aggregation. We then apply the method to characterize the complex formed by the histidine kinase CheA and its target response regulator CheY. The X-ray structure of CheY–PyTyr confirms Cu labeling at PyTyr but also reveals a secondary Cu site. Cu2+-to-NO and Cu2+-to-Cu2+ PDS measurements of CheY–PyTyr with nitroxide-labeled CheA provide new insights into the conformational landscape of the phosphotransfer complex and have implications for kinase regulation.
|
|
|
Phenyl-Ring Dynamics in Amyloid Fibrils and Proteins: The Microscopic-Order-Macroscopic-Disorder Perspective
E. Meirovitch, Z. Liang, and J. H. Freed.
J. Phys. Chem. B 122, 8675-8684 (2018)
Supporting Information
<doi: 10.1021/acs.jpcb.8b06330>
PMID:
30141954
PMCID:
PMC6174686
|
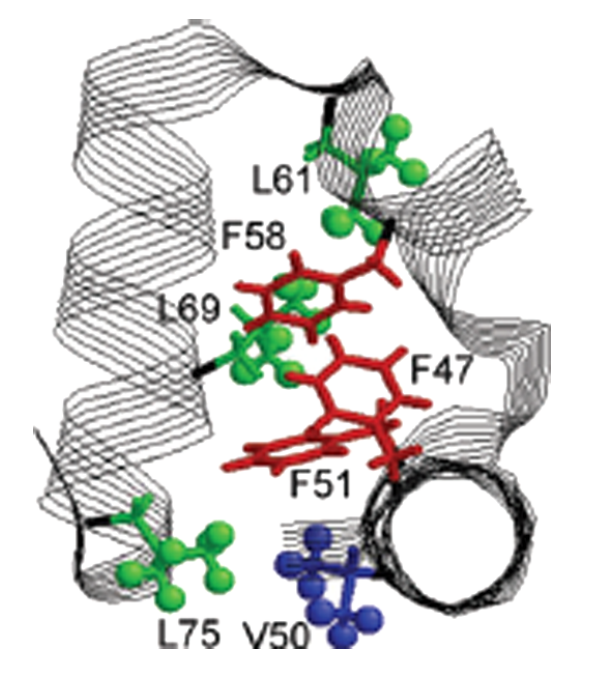
|
|
ABSTRACT: We have developed the microscopic-order-macroscopic-disorder (MOMD) approach for studying internal mobility in polycrystalline proteins with 2H lineshape analysis. The motion itself is expressed by a diffusion tensor, R, the local spatial restraints by a potential, u, and the "local geometry" by the relative orientation of the model-related and nuclear magnetic resonance-related tensors. Here, we apply MOMD to phenyl-ring dynamics in several Αβ40-amyloid-fibrils, and the villin headpiece subdomain (HP36). Because the available data are limited in extent and sensitivity, we adjust u and R in the relevant parameter ranges, fixing the "local geometry" in accordance with standard stereochemistry. This yields a physically well-defined and consistent picture of phenyl-ring dynamics, enabling comparison between different systems. In the temperature range of 278–308 K, u has a strength of (1.7–1.8) kT and a rhombicity of (2.4–2.6) kT, and R has components of 5.0 × 102 ≤ R⊥ ≤ 2.0 × 103 s–1 and 6.3 × 105 ≤ R∥ ≤ 2.0 × 106 s–1. At 278 K, fibril hydration increases the axiality of both u and R; HP36 hydration has a similar effect at 295 K, reducing R⊥ considerably. The D23N mutation slows down the motion of the probe; Αβ40 polymorphism affects both this motion and the related local potential. The present study identifies the impact of various factors on phenyl-ring mobility in amyloid fibrils and globular proteins; the difference between the two protein forms is considerable. The distinctive impact of hydration on phenyl-ring motion and previously studied methyl-group motion is also examined. The 2H lineshapes considered here were analyzed previously with various multi-simple-mode (MSM) models, where several simple motional modes are combined. The MOMD and MSM interpretations differ in essence.
|
|
|
Open and Closed Form of Maltose Binding Protein in Its Native and Molten Globule State As Studied by Electron Paramagnetic Resonance Spectroscopy
B. Selmke, P. P. Borbat, C. Nickolaus, R. Varadarajan, J. H. Freed, and W. E. Trommer.
Biochemistry 57, 5507-5512 (2018)
Supporting Information
<doi: 10.1021/acs.biochem.8b00322>
PMID:
30004675
PMCID:
PMC6211580
|
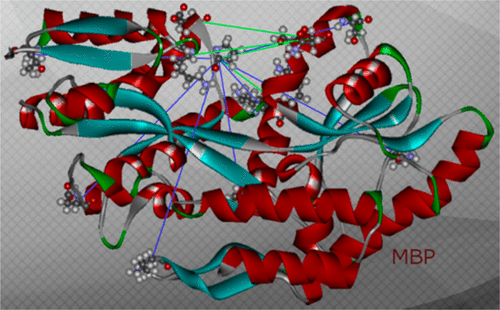
|
|
ABSTRACT: An intensively investigated intermediate state of protein folding is the molten globule (MG) state, which contains secondary but hardly any tertiary structure. In previous work, we have determined the distances between interacting spins within maltose binding protein (MBP) in its native state using continuous wave and double electron–electron resonance (DEER) electron paramagnetic resonance (EPR) spectroscopy. Seven double mutants had been employed to investigate the structure within the two domains of MBP. DEER data nicely corroborated the previously available X-ray data. Even in its MG state, MBP is known to still bind its ligand maltose. We therefore hypothesized that there must be a defined structure around the binding pocket of MBP already in the absence of tertiary structure. Here we have investigated the functional and structural difference between native and MG state in the open and closed form with a new set of MBP mutants. In these, the spin-label positions were placed near the active site. Binding of its ligands leads to a conformational change from open to closed state, where the two domains are more closely together. The complete set of MBP mutants was analyzed at pH 3.2 (MG) and pH 7.4 (native state) using double-quantum coherence EPR. The values were compared with theoretical predictions of distances between the labels in biradicals constructed by molecular modeling from the crystal structures of MBP in open and closed form and were found to be in excellent agreement. Measurements show a defined structure around the binding pocket of MBP in MG, which explains maltose binding. A new and important finding is that in both states ligand-free MBP can be found in open and closed form, while ligand-bound MBP appears only in closed form because of maltose binding.
|
|
|
A facile approach for the in vitro assembly of multimeric membrane transport proteins
E. A. Riederer, P. J. Focke, E. R. Georgieva, N. Akyuz, K. Matulef, P. P. Borbat, J. H. Freed, S. C. Blanchard, O. Boudker, and F. I. Valiyaveetil.
eLife 7, e36478 (2018)
|
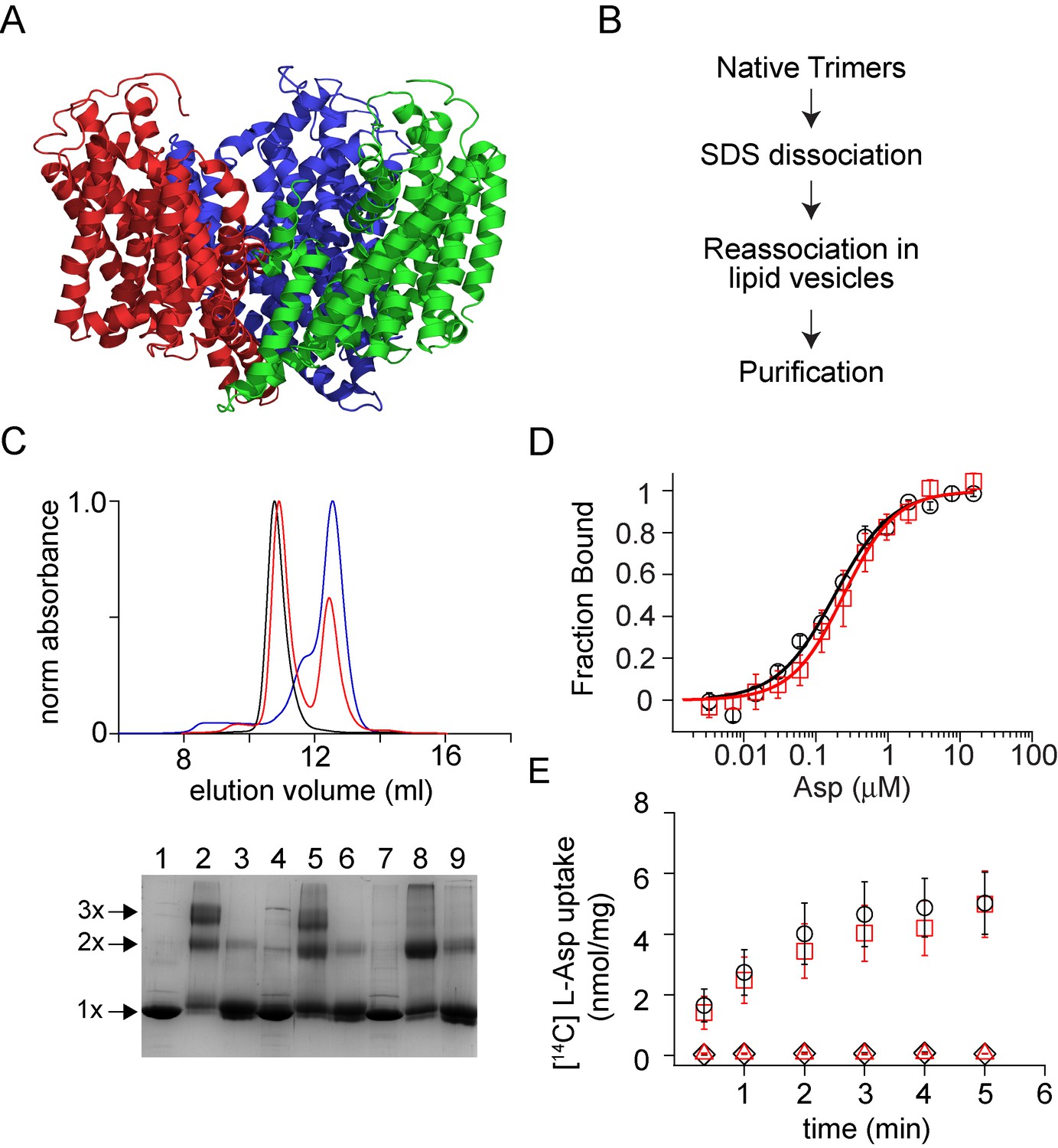
|
|
A facile approach for the in vitro assembly of multimeric membrane transport proteins
E. A. Riederer, P. J. Focke, E. R. Georgieva, N. Akyuz, K. Matulef, P. P. Borbat, J. H. Freed, S. C. Blanchard, O. Boudker, and F. I. Valiyaveetil.
eLife 7, e36478 (2018)
Supporting Information
<doi: 10.7554/eLife.36478>
PMID:
29889023
PMCID:
PMC6025958
|
|
|
ABSTRACT: Membrane proteins such as ion channels and transporters are frequently homomeric. The homomeric nature raises important questions regarding coupling between subunits and complicates the application of techniques such as FRET or DEER spectroscopy. These challenges can be overcome if the subunits of a homomeric protein can be independently modified for functional or spectroscopic studies. Here, we describe a general approach for in vitro assembly that can be used for the generation of heteromeric variants of homomeric membrane proteins. We establish the approach using GltPh, a glutamate transporter homolog that is trimeric in the native state. We use heteromeric GltPh transporters to directly demonstrate the lack of coupling in substrate binding and demonstrate how heteromeric transporters considerably simplify the application of DEER spectroscopy. Further, we demonstrate the general applicability of this approach by carrying out the in vitro assembly of VcINDY, a Na+-coupled succinate transporter and CLC-ec1, a Cl-/H+ antiporter.
|
|
|
Structural basis for membrane anchoring and fusion regulation of the herpes simplex virus fusogen gB
R. S. Cooper, E. R. Georgieva, P. P. Borbat, J. H. Freed, and E. E. Heldwein.
Nat. Struct. Mol. Biol. 25, 416-424 (2018)
Supporting Information
<doi: 10.1038/s41594-018-0060-6>
PMID:
29728654
PMCID:
PMC5942590
|
|
|
ABSTRACT: Viral fusogens merge viral and cell membranes during cell penetration. Their ectodomains drive fusion by undergoing large-scale refolding, but little is known about the functionally important regions located within or near the membrane. Here we report the crystal structure of full-length glycoprotein B (gB), the fusogen from herpes simplex virus, complemented by electron spin resonance measurements. The membrane-proximal (MPR), transmembrane (TMD), and cytoplasmic (CTD) domains form a uniquely folded trimeric pedestal beneath the ectodomain, which balances dynamic flexibility with extensive, stabilizing membrane interactions. The postfusion conformation of the ectodomain suggests that the CTD likewise adopted the postfusion form. However, hyperfusogenic mutations, which destabilize the prefusion state of gB, target key interfaces and structural motifs that reinforce the observed CTD structure. Thus, a similar CTD structure must stabilize gB in its prefusion state. Our data suggest a model for how this dynamic, membrane-dependent 'clamp' controls the fusogenic refolding of gB.
|
|
|
MOMD Analysis of NMR Line Shapes from Aβ-Amyloid Fibrils: A New Tool for Characterizing Molecular Environments in Protein Aggregates
E. Meirovitch, Z. Liang, and J. H. Freed.
J. Phys. Chem. B 122, 4793-4801 (2018)
Supporting Information
<doi: 10.1021/acs.jpcb.8b02181>
PMID:
29624402
PMCID:
PMC5945340
|
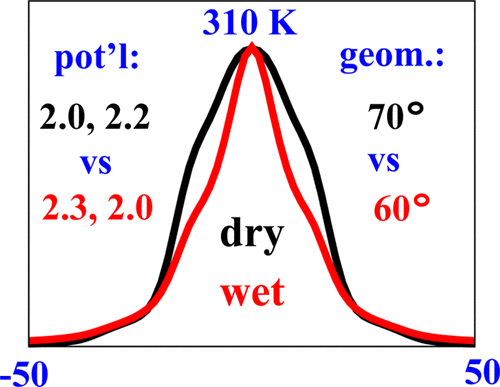
|
|
ABSTRACT: The microscopic-order-macroscopic-disorder (MOMD) approach for 2H NMR line shape analysis is applied to dry and hydrated 3-fold- and 2-fold-symmetric amyloid-Aβ40 fibrils and protofibrils of the D23N mutant. The methyl moieties of L17, L34, V36 (C–CD3), and M35 (S–CD3) serve as probes. Experimental 2H spectra acquired previously in the 147–310 K range are used. MOMD describes local probe motion as axial diffusion (R tensor) in the presence of a potential, u, which represents the spatial restrictions exerted by the molecular surroundings. We find that R∥ = (0.2–3.3) × 104 s–1, R⊥ = (2.2–2.5) × 102 s–1, and R is tilted from the 2H quadrupolar tensor at 60–75°. The strength of u is in the (2.0–2.4) kT range; its rhombicity is substantial. The only methyl moieties affected by fibril hydration are those of M35, located at fibril interfaces. The associated local potentials change form abruptly around 260 K, where massive water freezing occurs. An independent study revealed unfrozen "tightly-peptide-bound" water residing at the interfaces of the 3-fold-symmetric Aβ40 fibrils and at the interfaces of the E22G and E22Δ Aβ40-mutant fibrils. Considering this to be the case in general for Aβ40-related fibrils, the following emerges. The impact of water freezing is transmitted selectively to the fibril structure through interactions with tightly-peptide-bound water, in this case of M35 methyl moieties. The proof that such waters reside at the interfaces of the 2-fold-symmetric fibril, and the protofibril of the D23N mutant, is new. MOMD provides information on the surroundings of the NMR probe directly via the potential, u, which is inherent to the model; a prior interpretation of the same experimental data does so partially and indirectly (see below). Thus, MOMD analysis of NMR line shapes as applied to amyloid fibrils/protein aggregates emerges as a consistent new tool for elucidating the properties of, and processes associated with, molecular environments in the fibril.
|
|
|
Organometallic and radical intermediates reveal mechanism of diphthamide biosynthesis
M. Dong, V. Kathiresan, M. K. Fenwick, A. T. Torelli, Y. Zhang, J. D. Caranto, B. Dzikovski, A. Sharma, K. M. Lancaster, J. H. Freed, S. E. Ealick, B. M. Hoffman, and H. Lin.
Science 359, 1247-1250 (2018)
|
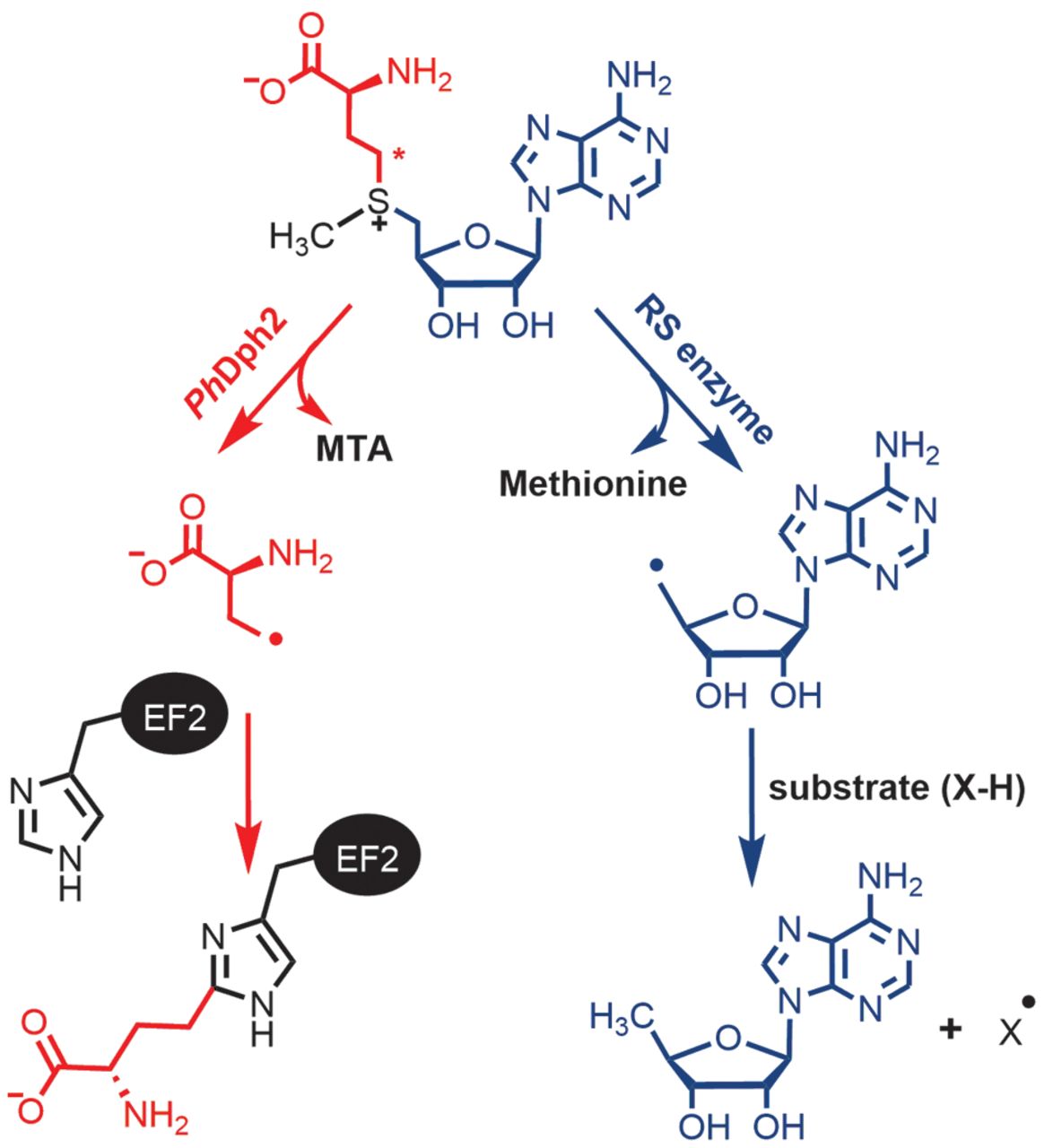
|
|
Organometallic and radical intermediates reveal mechanism of diphthamide biosynthesis
M. Dong, V. Kathiresan, M. K. Fenwick, A. T. Torelli, Y. Zhang, J. D. Caranto, B. Dzikovski, A. Sharma, K. M. Lancaster, J. H. Freed, S. E. Ealick, B. M. Hoffman, and H. Lin.
Science 359, 1247-1250 (2018)
Supporting Information
<doi: 10.1126/science.aao6595>
PMID:
29590073
PMCID:
PMC6066404
|
|
|
ABSTRACT: Diphthamide biosynthesis involves a carbon-carbon bond-forming reaction catalyzed by a radical S-adenosylmethionine (SAM) enzyme that cleaves a carbon-sulfur (C–S) bond in SAM to generate a 3-amino-3-carboxypropyl (ACP) radical. Using rapid freezing, we have captured an organometallic intermediate with an iron-carbon (Fe–C) bond between ACP and the enzyme's [4Fe-4S] cluster. In the presence of the substrate protein, elongation factor 2, this intermediate converts to an organic radical, formed by addition of the ACP radical to a histidine side chain. Crystal structures of archaeal diphthamide biosynthetic radical SAM enzymes reveal that the carbon of the SAM C–S bond being cleaved is positioned near the unique cluster Fe, able to react with the cluster. Our results explain how selective C–S bond cleavage is achieved in this radical SAM enzyme.
|
|
|
Protein dynamics in the solid-state from 2H NMR lineshape analysis. III. MOMD in the presence of Magic Angle Spinning
E. Meirovitch, Z. Liang, J. H. Freed.
Solid State Nucl. Magn. Reson. 89, 35-44 (2018)
<doi: 10.1016/j.ssnmr.2017.11.001>
PMID:
29208317
PMCID:
PMC5772661
|
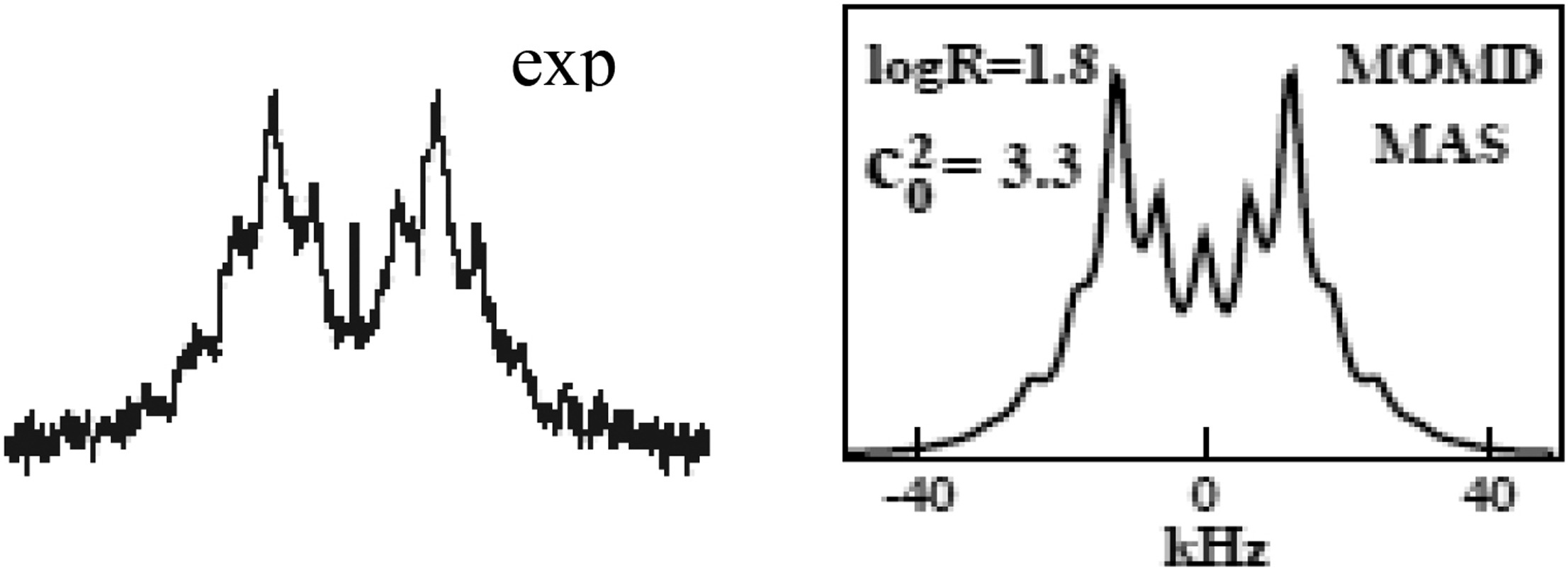
|
|
ABSTRACT: We report on a new approach to the analysis of dynamic NMR lineshapes from polycrystalline (i.e., macroscopically disordered) samples in the presence of Magic Angle Spinning (MAS). This is an application of the Stochastic Liouville Equation developed by Freed and co-workers for treating restricted (i.e., microscopically ordered) motions. The 2H nucleus in an internally-mobile C–CD3 moiety serves as a prototype probe. The acronym is 2H/MOMD/MAS, where MOMD stands for "microscopic-order-macroscopic-disorder." The key elements describing internal motions – their type, the local spatial restrictions, and related features of local geometry – are treated in MOMD generally, within their rigorous three-dimensional tensorial requirements. Based on this representation a single physically well-defined model of local motion has the capability of reproducing experimental spectra. There exist other methods for analyzing dynamic 2H/MAS spectra which advocate simple motional modes. Yet, to reproduce satisfactorily the experimental lineshapes, one has either to use unusual parameter values, or combine several simple motional modes. The multi-simple-mode reasoning assumes independence of the constituent modes, features ambiguity as different simple modes may be used, renders inter-system comparison difficult as the overall models differ, and makes possible model-improvement only by adding yet another simple mode, i.e., changing the overall model. 2H/MOMD/MAS is free of such limitations and inherently provides a clear physical interpretation. These features are illustrated. The advantage of 2H/MOMD/MAS in dealing with sensitive but hardly investigated slow-motional lineshapes is demonstrated by applying it to actual experimental data. The results differ from those obtained previously with a two-site exchange scheme that yielded unusual parameters.
|
|
© 2022
|
.svg)