|
Demo of new pubs function
A. L. Lai, N, K. Singhota, D. E. C. Patzer
Journal of Irreproducible Results
|
|
|
Demo of new pubs function
A. L. Lai, N, K. Singhota, D. E. C. Patzer
Journal of Irreproducible Results
|
|
|
ABSTRACT: Just playing with the system to test functions.
|
|
|
Flavoproteins as native and genetically encoded spin probes for in cell ESR spectroscopy
T. Chauviré, S. Chandrasekaran, R. Dunleavy, J. H. Freed, B. R. Crane
Nat. Commun. 16, 5406 (2025).
Supporting Information
<doi: 10.1038/s41467-025-60623-6>
PMID:
40595551
PMCID:
PMC12214676
|
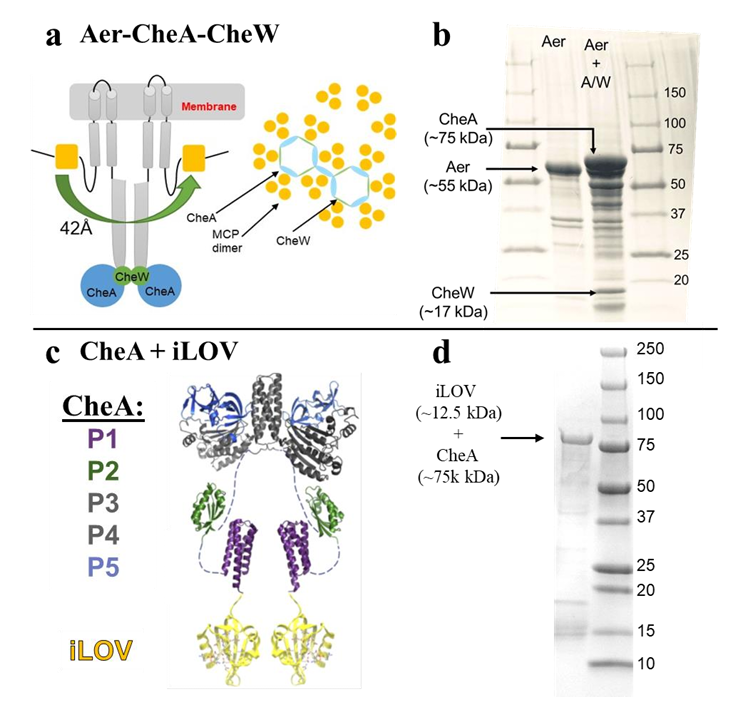
|
|
ABSTRACT: Flavin cofactors are attractive Electron Spin Resonance (ESR) probes for proteins because cellular reductants and light can generate their semiquinone states. We have used ESR spectroscopy to study the bacterial transmembrane aerotaxis receptor (Aer) in its native Escherichia coli membrane environment. Optimization of the spectroscopic (electronic relaxation times) and cell growth (isotopic labeling) conditions allowed for measurements of Aer with its partners - the histidine kinase (CheA) and the coupling protein (CheW) - in native signaling arrays. Continuous-wave ESR measurements at room temperature showed a rigid Aer flavin immobilized in the cofactor pocket and Q-band electron nuclear double resonance (ENDOR) measurements identified a predominant anionic semiquinone radical state in cell. Q-band four-pulse double electron-electron resonance (4P-DEER) measurements indicated a 4.1 nm distance between the two flavins of an Aer homodimer, consistent with previous in vitro measurements, but also revealed additional separations in cell indicative of chemoreceptor arrays, not previously observed for Aer. For general application, we further developed a genetically encoded Light-Oxygen and Voltage (LOV) domain for incorporation into target proteins as an ESR probe of structural properties in cell. This approach provides a framework to elucidate protein oligomeric states and conformations that are difficult to reproduce in vitro.
|
|
|
pH-Modulated activation of a pendant amine leading to rapid electrocatalytic H2 production by a molecular copper complex in acidic water
N. A. Shah, T. Dolkar, S. Karim, J. Ishrat, C. Das, S. Das, A. Sinha Roy, K. Bhattacharyya, and A. Dutta
Inorg. Chem. Front. (2025) Advance Article
|
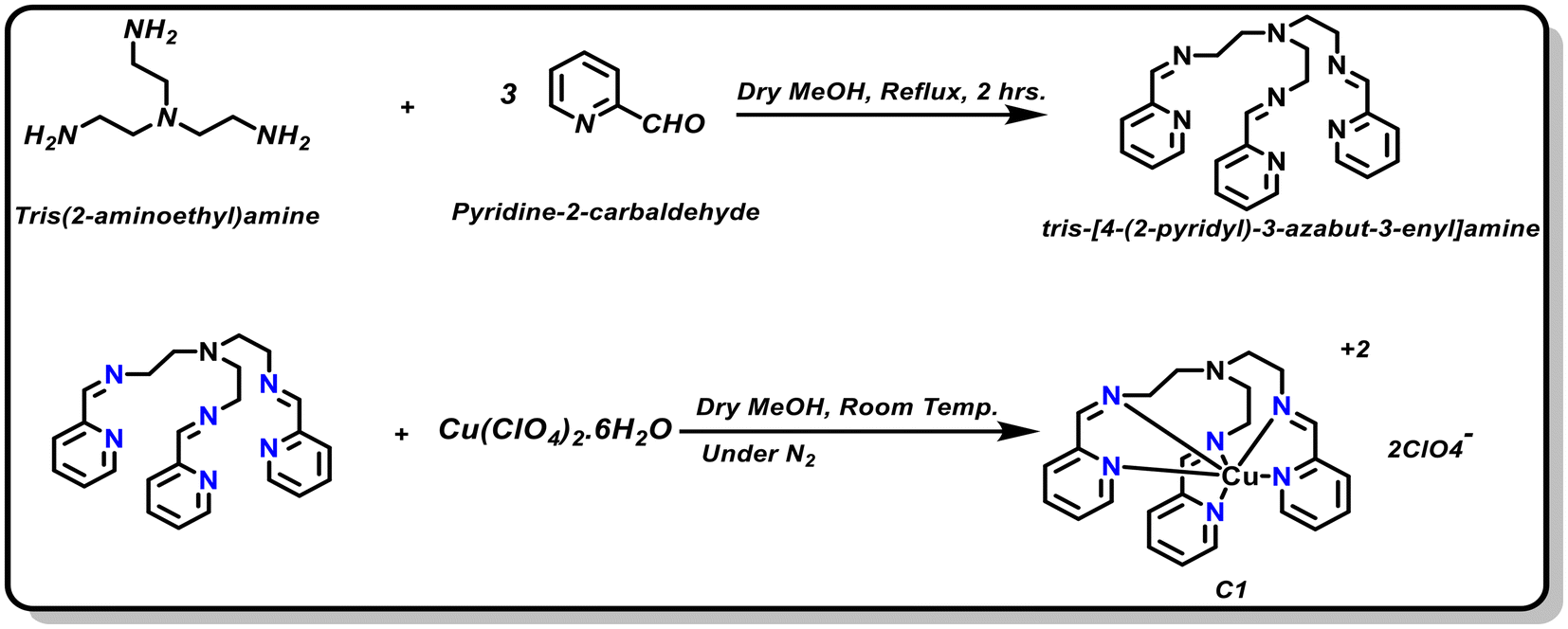
|
|
pH-Modulated activation of a pendant amine leading to rapid electrocatalytic H2 production by a molecular copper complex in acidic water
N. A. Shah, T. Dolkar, S. Karim, J. Ishrat, C. Das, S. Das, A. Sinha Roy, K. Bhattacharyya, and A. Dutta
Inorg. Chem. Front. (2025) Advance Article
<doi: 10.1039/D5QI00963D>
|
|
|
ABSTRACT: A modular multidentate ligand scaffold is crafted by strategically incorporating three pyridines (NPy) and three imines along with a pendant tertiary amine (Ntert) around a mononuclear copper centre. This unique design leads to the generation of a molecular copper complex C1 with a dynamically adaptive coordination environment, where multiple proton and electron movements can be accommodated. Complex C1 demonstrates rapid hydrogen generation from water across a wide pH range (pH 1.0−7.0), with a markedly enhanced catalytic performance under acidic conditions. At pH 1.0, C1 achieves high turnover numbers (TONs) of 1014 ± 10 within 1 hour and 2980 ± 20 over 3 hours. In operando spectroelectrochemical investigations, in conjunction with density functional theory (DFT) calculations, reveal a unique pH-dependent structural flexibility of the ligand scaffold around the Cu centre in C1. In near-neutral to slightly acidic media (pH 3.0−7.0), the protonation of an NPy group (pKa1 ˜ 11.6) following its cleavage from the Cu linkage provides the primary protonation site, which is essential for Cu-complex-driven H2 production catalysis. The Ntert group (pKa2 ˜ 2.8), positioned in the outer coordination sphere of Cu, becomes involved under highly acidic conditions (pH < 3.0). Here, this pendant amine acts as the initial protonation site and alters the course of the catalysis by unleashing an energetically downhill reaction pathway consisting of spontaneous electron and proton transfer steps. This pH-specific participation of the pendant Ntert functionality is key for the escalated HER activity by C1 under strongly acidic conditions, which is rarely observed for Cu-based molecular complexes. Complementary surface and solution-phase analyses confirm the molecular integrity of the complex, supporting a homogeneous catalytic mechanism operative throughout the hydrogen evolution process.
|
|
|
Halogenated Cholesterol Alters the Phase Behavior of Ternary Lipid Membranes
D. Mehta, E. K. Crumley, J. Lou, B. Dzikovski, M. D. Best, M. N. Waxham, F. A. Heberle
J. Phys. Chem. B 129, (2) 671-683 (2025)
|
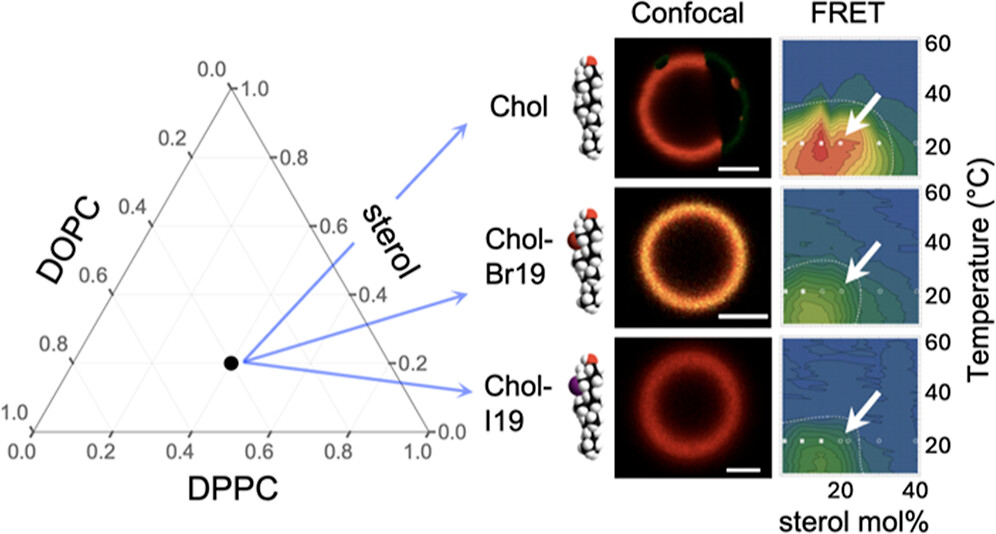
|
|
Halogenated Cholesterol Alters the Phase Behavior of Ternary Lipid Membranes
D. Mehta, E. K. Crumley, J. Lou, B. Dzikovski, M. D. Best, M. N. Waxham, F. A. Heberle
J. Phys. Chem. B 129, (2) 671-683 (2025)
<doi: 10.1021/acs.jpcb.4c06318>
PMID:
39772574
|
|
|
ABSTRACT: Eukaryotic plasma membranes exhibit nanoscale lateral lipid heterogeneity, a feature that is thought to be central to their function. Studying these heterogeneities is challenging since few biophysical methods are capable of detecting domains at submicron length scales. We recently showed that cryogenic electron microscopy (cryo-EM) can directly image nanoscale liquid–liquid phase separation in extruded liposomes due to its ability to resolve the intrinsic thickness and electron density differences of ordered and disordered phases. However, the intensity contrast between these phases is poor compared with conventional fluorescence microscopy and is thus both a limiting factor and a focal point for optimization. Because the fundamental source of intensity contrast is the spatial variation in electron density within the bilayer, lipid modifications aimed at selectively increasing the electron density of one phase might enhance the ability to resolve coexisting phases. To this end, we investigated model membrane mixtures of DPPC/DOPC/cholesterol in which one hydrogen of cholesterol's C19 methyl group was replaced by an electron-rich halogen atom (either bromine or iodine). We characterized the phase behavior as a function of composition and temperature using fluorescence microscopy, Förster resonance energy transfer, and cryo-EM. Our data suggest that halogenated cholesterol variants distribute approximately evenly between liquid-ordered and liquid-disordered phases and are thus ineffective at enhancing the intensity difference between them. Furthermore, replacing more than half of the native cholesterol with halogenated cholesterol variants dramatically reduces the size of the membrane domains. Our results reinforce how small changes in the sterol structure can have a large impact on the lateral organization of membrane lipids.
|
|
|
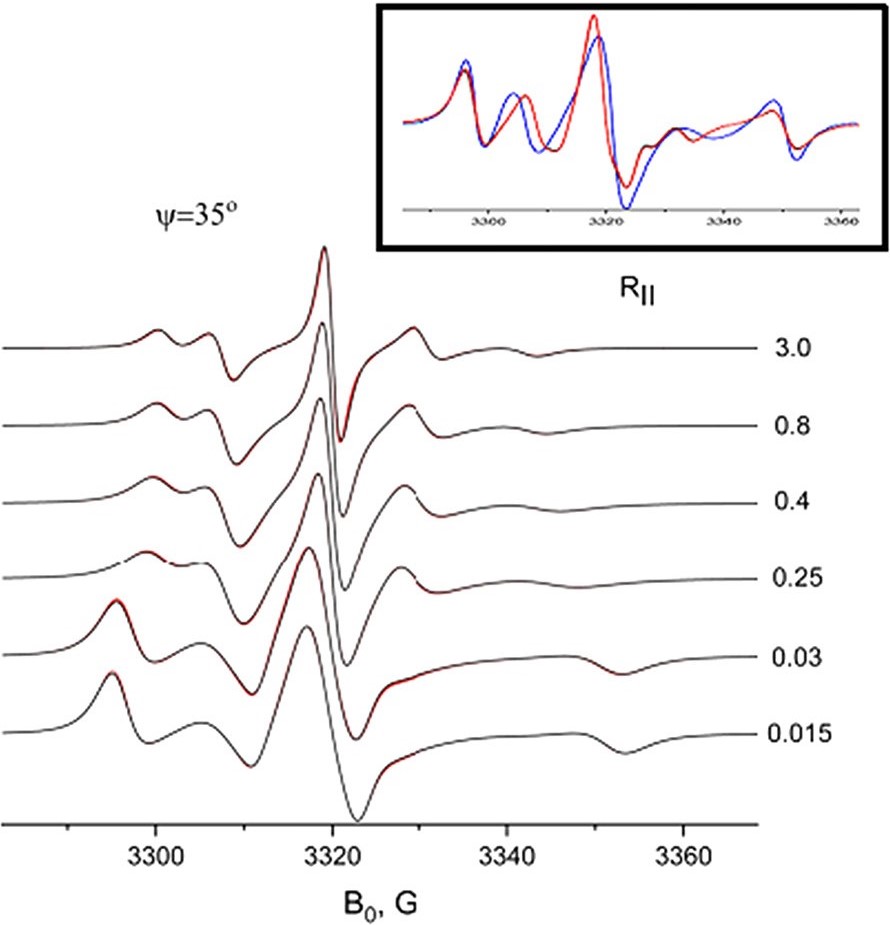
Rapid Analysis of DEER Signals Including Short Distances
A. Sinha Roy, T. E. Assafa, B. Dzikovski, N. Joshi, J. H. Freed
J. Phys. Chem. Lett. 16, (1) 38-44 (2025)
|
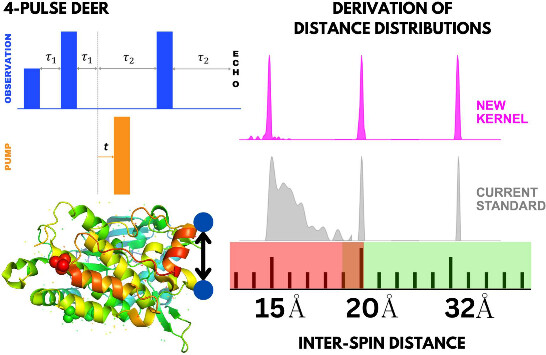
|
|
Rapid Analysis of DEER Signals Including Short Distances
A. Sinha Roy, T. E. Assafa, B. Dzikovski, N. Joshi, J. H. Freed
J. Phys. Chem. Lett. 16, (1) 38-44 (2025)
<doi: 10.1021/acs.jpclett.4c03245>
PMID:
39693563
PMCID:
PMC11717586
|
|
|
ABSTRACT: Double electron electron resonance (DEER) spectroscopy is an important technique to measure distance distributions P(r) for studying protein structures and protein–protein interactions. DEER data analysis can at times become challenging due to the lack of a detailed analytical signal expression or numerical methods with rapid computation time. We have derived an analytical expression κFULL, which includes both the pseudo-secular dipolar coupling (PSDC) and the finite pulse effects, especially important for shorter distances. Analyses of experiments by κFULL yield accurate and consistent P(r) values for three DEER nitroxide-rulers with distances (rAVG) in the range of 15 to 32 Å, while the current standard analysis produces erroneous results for rAVG < 20 Å. Computation times for deriving P(r) vary between 1 min and 4 min, which is usually much shorter than previous methods that include pseudo-secular and other effects. The expression can be applied to all types of DEER spin probes with little or no modifications.
|
|
|
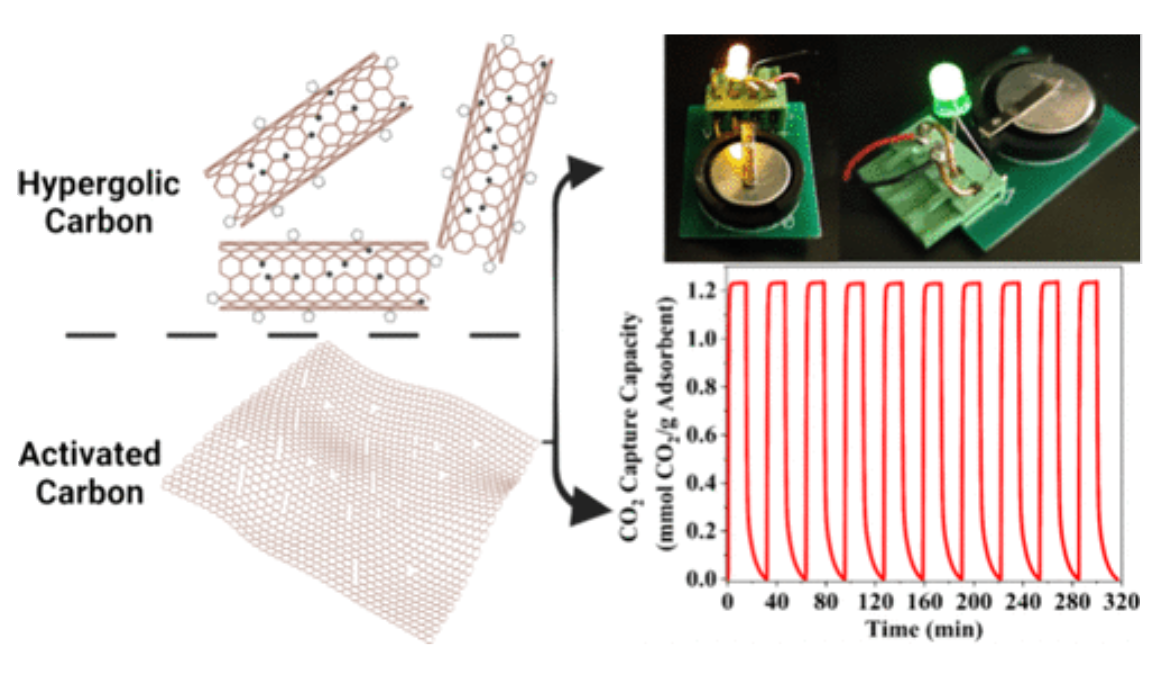
Ultrahigh Surface Area Nanoporous Carbons Synthesized via Hypergolic and Activation Reactions for Enhanced CO2 Capacity and Volumetric Energy Density
N. Chalmpes, P. Ochonma, I. Tantis, A. W. Alsmaeil, T. E. Assafa, M. Tathacharya, M. Srivastava, G. Gadikota, A. B. Bourlinos, T. Steriotis, E. P. Giannelis
ACS Nano 18 33491–33504 (2024)
|
|
|
Ultrahigh Surface Area Nanoporous Carbons Synthesized via Hypergolic and Activation Reactions for Enhanced CO2 Capacity and Volumetric Energy Density
N. Chalmpes, P. Ochonma, I. Tantis, A. W. Alsmaeil, T. E. Assafa, M. Tathacharya, M. Srivastava, G. Gadikota, A. B. Bourlinos, T. Steriotis, E. P. Giannelis
ACS Nano 18 33491–33504 (2024)
<doi: 10.1021/acsnano.4c10531>
PMID:
39576877
PMCID:
[In Progress]
|
|
|
ABSTRACT: We report a family of carbon sorbents synthesized by integrating hypergolics with activation reactions on a templated substrate. The materials design leads to nanoporous carbons with a BET area of 4800 m2 g–1 with an impressive total pore volume of 2.7 cm3 g–1. To the best of our knowledge, this BET area value is the highest reported in the literature. Electron spin resonance (ESR) measurements determined the number of radicals in an effort to provide a mechanistic understanding of the formation of ultrahigh surface area carbons. In combination with XPS, we propose a mechanism based on the synergistic effect between rim-based pentagonal rings and carbon radicals, which we believe can be exploited to produce other highly porous carbons. The CO2 capture capacity of the hyperporous carbon tested under dynamic CO2 capture conditions was ˜1.25 mmol g–1 versus 0.66 mmol g–1 of a conventionally activated carbon under similar conditions. The CO2 capture kinetics were extremely fast and reached 99% of the total capacity within 120 s. Lastly, supercapacitor electrodes deliver a high volumetric energy density of ˜60 W h L–1 and a volumetric power density of 1 kW L–1, which is the highest reported value for activated carbon.
|
|
|
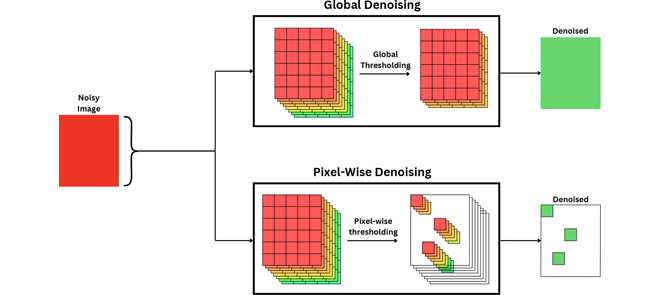
MRI Denoising Using Pixel-Wise Threshold Selection
N. Srivastava, G.R. Sahoo, H. U. Voss, S. N. Niogi, J. H. Freed, and M. Srivastava
IEEE Access 12 135730-135745 (2024)
<doi: 10.1109/ACCESS.2024.3449811>
PMID:
39640512
PMCID:
PMC11619618
|
|
|
ABSTRACT: Magnetic resonance imaging (MRI) has emerged as a promising technique for non-invasive medical imaging. The primary challenge in MRI is the trade-off between image visual quality and acquisition time. Current MRI image denoising algorithms employ global thresholding to denoise the whole image, which leads to inadequate denoising or image distortion. This study introduces a novel pixel-wise (localized) thresholding approach of singular vectors, obtained from singular value decomposition, to denoise magnetic resonance (MR) images. The pixel-wise thresholding of singular vectors is performed using separate singular values as thresholds at each pixel, which is advantageous given the spatial noise variation throughout the image. The method presented is validated on MR images of a standard phantom approved by the magnetic resonance accreditation program (MRAP). The denoised images display superior visual quality and recover minute structural information otherwise suppressed in the noisy image. The increase in peak-signal-to-noise-ratio (PSNR) and contrast-to-noise-ratio (CNR) values of ≥ 18% and ≥ 200% of the denoised images, respectively, imply efficient noise removal and visual quality enhancement. The structural similarity index (SSIM) of ≥ 0.95 for denoised images indicates that the crucial structural information is recovered through the presented method. A comparison with the standard filtering methods widely used for MRI denoising establishes the superior performance of the presented method. The presented pixel-wise denoising technique reduces the scan time by 2-3 times and has the potential to be integrated into any MRI system to obtain faster and better quality images.
|
|
|
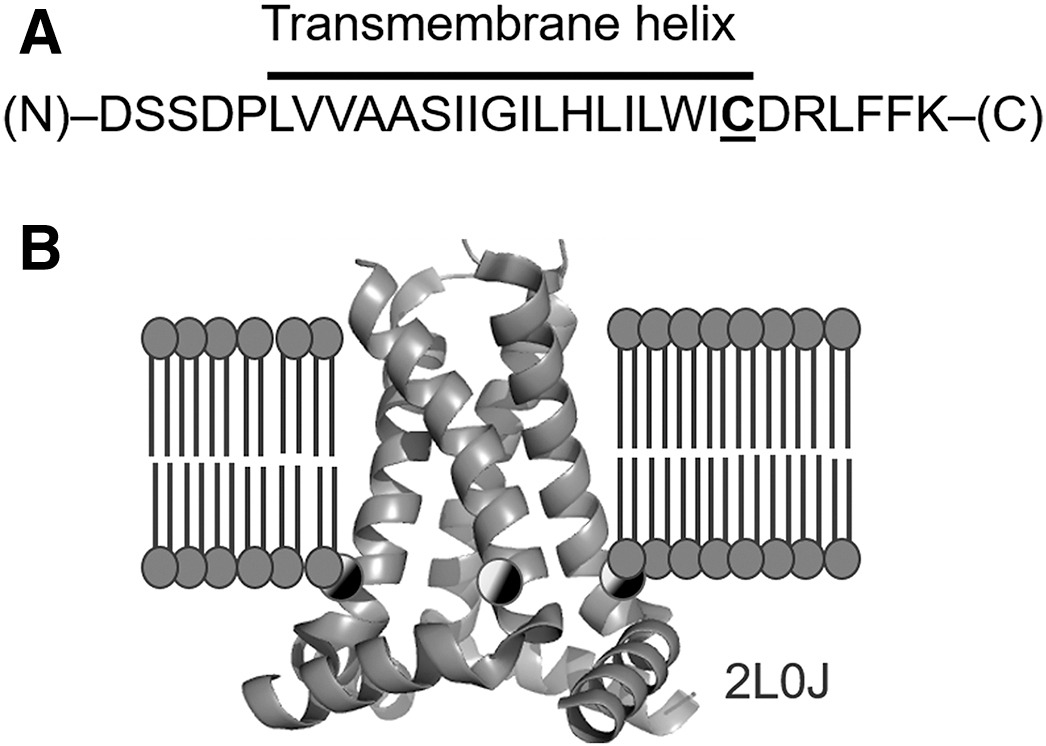
Conformations of influenza A M2 protein in DOPC/DOPS and E. coli native lipids and proteins
G. Sanders, P. P. Borbat, E. R. Georgieva
Biophys. J. 123, 2584-2593 (2024)
|
|
|
Conformations of influenza A M2 protein in DOPC/DOPS and E. coli native lipids and proteins
G. Sanders, P. P. Borbat, E. R. Georgieva
Biophys. J. 123, 2584-2593 (2024)
<doi: 10.1016/j.bpj.2024.06.025>
PMID:
38932458
PMCID:
PMC11365223
|
|
|
ABSTRACT: We compared the conformations of the transmembrane domain (TMD) of influenza A M2 (IM2) protein reconstituted in 1,2-dioleoyl-sn-glycero-3-phosphocholine/1,2-dioleoyl-sn-glycero-3-phospho-L-serine (DOPC/DOPS) bilayers to those in isolated Escherichia coli (E. coli) membranes, having preserved its native proteins and lipids. IM2 is a single-pass transmembrane protein known to assemble into a homo-tetrameric proton channel. To represent this channel, we made a construct containing the IM2's TMD region flanked by the juxtamembrane residues. The single cysteine substitution, L43C, of leucine located in the bilayer polar region was paramagnetically tagged with a methanethiosulfonate nitroxide label for the electron spin resonance (ESR) study. For this particular residue, we probed the conformations of the spin-labeled IM2 reconstituted in DOPC/DOPS and isolated E. coli membranes using continuous-wave ESR and double electron-electron resonance (DEER) spectroscopy. The total protein-to-lipid molar ratio spanned the range from 1:230 to 1:10,400. The continuous-wave ESR spectra corresponded to very slow spin-label motion in both environments. In all cases, the DEER data were reconstructed into distance distributions with well-resolved peaks at 1.68 and 2.37 nm in distance and amplitude ratios of 1.41 ± 0.2 and 2:1, respectively. This suggests four nitroxide spin labels located at the corners of a square, indicative of an axially symmetric tetramer. The distance modeling of DEER data with molecular modeling software applied to the NMR molecular structures (PDB: 2L0J) confirmed the symmetry and closed state of the C-terminal exit pore of the IM2 TMD tetramer in agreement with the model. Thus, we can conclude that, under conditions of pH 7.4 used in this study, IM2 TMD has similar conformations in model lipid bilayers and membranes made of native E. coli lipids and proteins of comparable thickness and fluidity, notwithstanding the complexity of the E. coli membranes caused by their lipid diversity and the abundance of integral and peripheral membrane proteins.
|
|
|
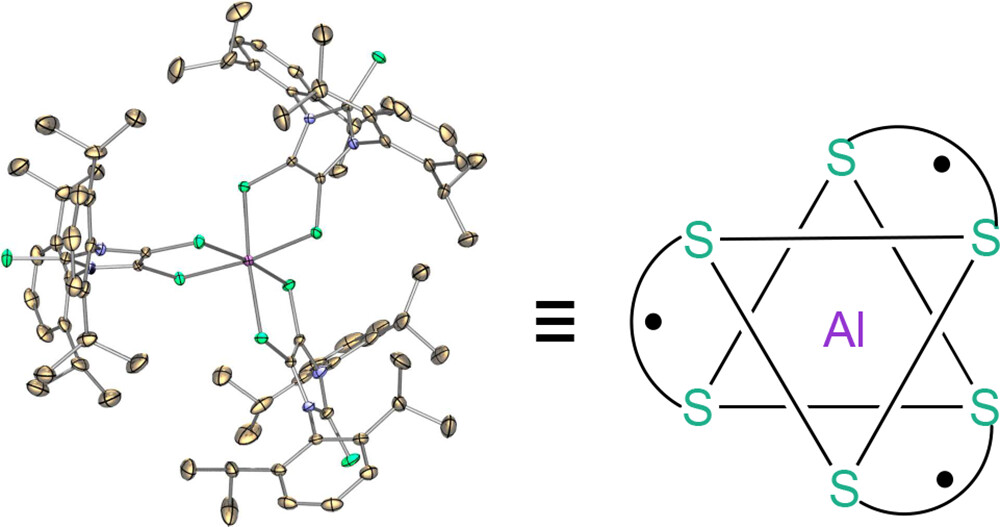
A Stable Aluminum Tris(dithiolene) Triradical
P. M. Tran, Y. Wang, B. Dzikovski, M. E. Lahm, Y. Xie, P. Wei, V. V. Klepov, H. F. Schaefer 3rd, G. H. Robinson
J. Am. Chem. Soc. 146, (23) 16340-16347 (2024)
Supporting Information
<doi: 10.1021/jacs.4c05631>
PMID:
38820231
PMCID:
PMC11177253
|
|
|
ABSTRACT: A stable aluminum tris(dithiolene) triradical (3) was experimentally realized through a low-temperature reaction of the sterically demanding lithium dithiolene radical (2) with aluminum iodide. Compound 3 was characterized by single-crystal X-ray diffraction, UV–vis and EPR spectroscopy, SQUID magnetometry, and theoretical computations. The quartet ground state of triradical 3 has been unambiguously confirmed by variable-temperature continuous wave EPR experiments and SQUID magnetometry. Both SQUID magnetometry and broken-symmetry DFT computations reveal a small doublet–quartet energy gap [ΔEDQ = 0.18 kcal mol–1 (SQUID); ΔEDQ = 0.14 kcal mol–1 (DFT)]. The pulsed EPR experiment (electron spin echo envelop modulation) provides further evidence for the interaction of these dithiolene-based radicals with the central aluminum nucleus of 3.
|
|
|
Novel requirements for HAP2/GCS1-mediated gamete fusion in Tetrahymena
J. F. Pinello, J. Loidl, E. S. Seltzer, D. Cassidy-Hanley, D. Kolbin, A. Abdelatif, F. A. Rey, R. An, N. J. Newberger, Y. Bisharyan, H. Papoyan, H. Byun, H. C. Aguilar, A. L. Lai, J. H. Freed, T. Maugel, E. S. Cole, and T. G. Clark
iScience 27, 110146 (2024)
|

|
|
Novel requirements for HAP2/GCS1-mediated gamete fusion in Tetrahymena
J. F. Pinello, J. Loidl, E. S. Seltzer, D. Cassidy-Hanley, D. Kolbin, A. Abdelatif, F. A. Rey, R. An, N. J. Newberger, Y. Bisharyan, H. Papoyan, H. Byun, H. C. Aguilar, A. L. Lai, J. H. Freed, T. Maugel, E. S. Cole, and T. G. Clark
iScience 27, 110146 (2024)
Supporting Information
<doi: 10.1016/j.isci.2024.110146>
PMID:
38904066
PMCID:
PMC11187246
|
|
|
ABSTRACT: The ancestral gamete fusion protein, HAP2/GCS1, plays an essential role in fertilization in a broad range of taxa. To identify factors that may regulate HAP2/GCS1 activity, we screened mutants of the ciliate Tetrahymena thermophila for behaviors that mimic Δhap2/gcs1 knockout phenotypes in this species. Using this approach, we identified two new genes, GFU1 and GFU2, whose products are necessary for membrane pore formation following mating type recognition and adherence. GFU2 is predicted to be a single-pass transmembrane protein, while GFU1, though lacking obvious transmembrane domains, has the potential to interact directly with membrane phospholipids in the cytoplasm. Like Tetrahymena HAP2/GCS1, expression of GFU1 is required in both cells of a mating pair for efficient fusion to occur. To explain these bilateral requirements, we propose a model that invokes cooperativity between the fusion machinery on apposed membranes of mating cells and accounts for successful fertilization in Tetrahymena's multiple mating type system.
|
|
|
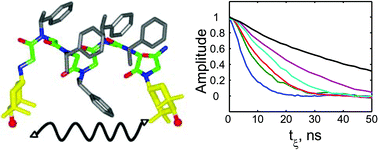
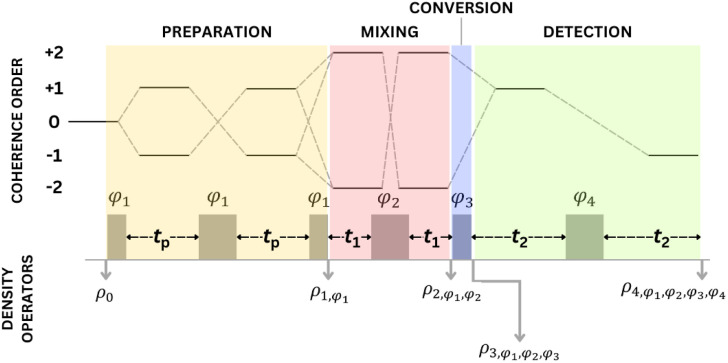
An analysis of double-quantum coherence ESR in an N-spin system: Analytical expressions and predictions
A. Sinha Roy, J. A. Marohn, J. H. Freed
J. Chem. Phys. 160, 134105 (2024)
|
|
|
An analysis of double-quantum coherence ESR in an N-spin system: Analytical expressions and predictions
A. Sinha Roy, J. A. Marohn, J. H. Freed
J. Chem. Phys. 160, 134105 (2024)
<doi: 10.1063/5.0200054>
PMID:
38557852
PMCID:
PMC11087869
|
|
|
ABSTRACT: Electron spin resonance pulsed dipolar spectroscopy (PDS) has become popular in protein 3D structure analysis. PDS studies yield distance distributions between a pair or multiple pairs of spin probes attached to protein molecules, which can be used directly in structural studies or as constraints in theoretical predictions. Double-quantum coherence (DQC) is a highly sensitive and accurate PDS technique to study protein structures in the solid state and under physiologically relevant conditions. In this work, we have derived analytical expressions for the DQC signal for a system with N-dipolar coupled spin-1/2 particles in the solid state. The expressions are integrated over the relevant spatial parameters to obtain closed form DQC signal expressions. These expressions contain the concentration-dependent "instantaneous diffusion" and the background signal. For micromolar and lower concentrations, these effects are negligible. An approximate analysis is provided for cases of finite pulses. The expressions obtained in this work should improve the analysis of DQC experimental data significantly, and the analytical approach could be extended easily to a wide range of magnetic resonance phenomena.
|
|
|
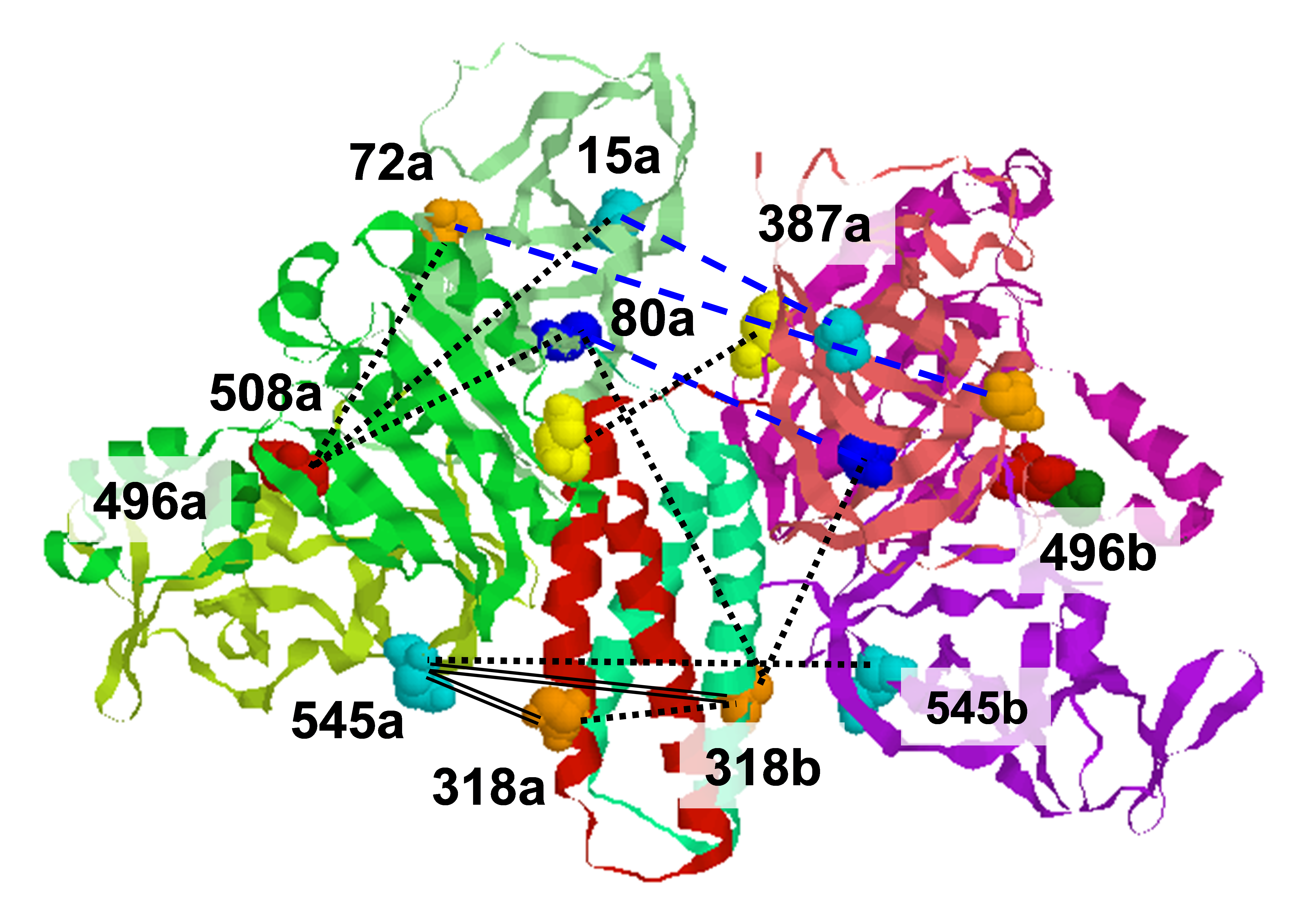
The crystal structure of bacteriophage λ RexA provides novel insights into the DNA binding properties of Rex-like phage exclusion proteins
M. C. Adams, C. J. Schiltz, J. Sun, C. J. Hosford, V. M. Johnson, H. Pan, P. P. Borbat, J. H. Freed, L. C. Thomason, C. Court, D. L. Court, J. S. Chappie
Nucleic Acids Res. 52, 4659-4675 (2024)
|

|
|
The crystal structure of bacteriophage λ RexA provides novel insights into the DNA binding properties of Rex-like phage exclusion proteins
M. C. Adams, C. J. Schiltz, J. Sun, C. J. Hosford, V. M. Johnson, H. Pan, P. P. Borbat, J. H. Freed, L. C. Thomason, C. Court, D. L. Court, J. S. Chappie
Nucleic Acids Res. 52, 4659-4675 (2024)
Supporting Information
<doi: 10.1093/nar/gkae212>
PMID:
38554102
PMCID:
PMC11077077
|
|
|
ABSTRACT: RexA and RexB function as an exclusion system that prevents bacteriophage T4rII mutants from growing on Escherichia coli λ phage lysogens. Recent data established that RexA is a non-specific DNA binding protein that can act independently of RexB to bias the λ bistable switch toward the lytic state, preventing conversion back to lysogeny. The molecular interactions underlying these activities are unknown, owing in part to a dearth of structural information. Here, we present the 2.05-Å crystal structure of the λ RexA dimer, which reveals a two-domain architecture with unexpected structural homology to the recombination-associated protein RdgC. Modelling suggests that our structure adopts a closed conformation and would require significant domain rearrangements to facilitate DNA binding. Mutagenesis coupled with electromobility shift assays, limited proteolysis, and double electron–electron spin resonance spectroscopy support a DNA-dependent conformational change. In vivo phenotypes of RexA mutants suggest that DNA binding is not a strict requirement for phage exclusion but may directly contribute to modulation of the bistable switch. We further demonstrate that RexA homologs from other temperate phages also dimerize and bind DNA in vitro. Collectively, these findings advance our mechanistic understanding of Rex functions and provide new evolutionary insights into different aspects of phage biology.
|
|
|
Phosphorylation, disorder, and phase separation govern the behavior of Frequency in the fungal circadian clock
D. Tariq, N. Maurici, B. M. Bartholomai, S. Chandrasekaran, J. C. Dunlap, A. Bah, B. R. Crane
eLife 12 RP90259 (2024)
Supporting Information
<doi: 10.7554/eLife.90259>
PMID:
38526948
PMCID:
PMC10963029
|

|
|
ABSTRACT: Circadian clocks are composed of transcription-translation negative feedback loops that pace rhythms of gene expression to the diurnal cycle. In the filamentous fungus Neurospora crassa, the proteins Frequency (FRQ), the FRQ-interacting RNA helicase (FRH), and Casein-Kinase I (CK1) form the FFC complex that represses expression of genes activated by the white-collar complex (WCC). FRQ orchestrates key molecular interactions of the clock despite containing little predicted tertiary structure. Spin labeling and pulse-dipolar electron spin resonance spectroscopy provide domain-specific structural insights into the 989-residue intrinsically disordered FRQ and the FFC. FRQ contains a compact core that associates and organizes FRH and CK1 to coordinate their roles in WCC repression. FRQ phosphorylation increases conformational flexibility and alters oligomeric state, but the changes in structure and dynamics are non-uniform. Full-length FRQ undergoes liquid–liquid phase separation (LLPS) to sequester FRH and CK1 and influence CK1 enzymatic activity. Although FRQ phosphorylation favors LLPS, LLPS feeds back to reduce FRQ phosphorylation by CK1 at higher temperatures. Live imaging of Neurospora hyphae reveals FRQ foci characteristic of condensates near the nuclear periphery. Analogous clock repressor proteins in higher organisms share little position-specific sequence identity with FRQ; yet, they contain amino acid compositions that promote LLPS. Hence, condensate formation may be a conserved feature of eukaryotic clocks.
|
|
|
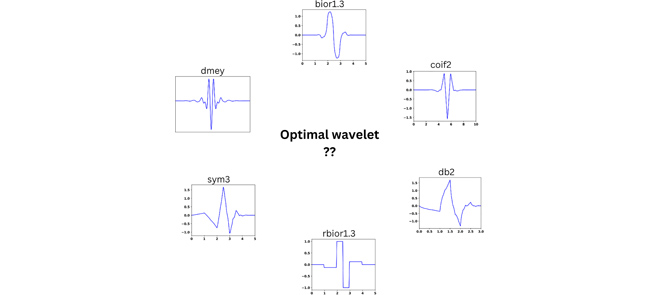
Optimal Wavelet Selection for Signal Denoising
G. R. Sahoo, J. H. Freed, M. Srivastava
IEEE Access 12 45369-45380 (2024)
<doi: 10.1109/ACCESS.2024.3377664>
PMID:
39421805
PMCID:
PMC11486496
|
|
|
ABSTRACT: Wavelet denoising plays a key role in removing noise from signals and is widely used in many applications. In denoising, selection of the mother wavelet is desirable for maximizing the separation of noise and signal coefficients in the wavelet domain for effective noise thresholding. At present, wavelet selection is carried out in a heuristic manner or using a trial-and-error that is time consuming and prone to error, including human bias. This paper introduces a universal method to select optimal wavelets based on the sparsity of Detail components in the wavelet domain, an empirical approach. A mean of sparsity change ( μsc ) parameter is defined that captures the mean variation of noisy Detail components. The efficacy of the presented method is tested on simulated and experimental signals from Electron Spin Resonance spectroscopy at various SNRs. The results reveal that the μsc values of signal vary abruptly between wavelets, whereas for noise it displays similar values for all wavelets. For low Signal-to-Noise Ratio (SNR) data, the change in μsc between highest and second highest value is ≈8–10% and for high SNR data it is around 5%. The mean of sparsity change increases with the SNR of the signal, which implies that multiple wavelets can be used for denoising a signal, whereas, the signal with low SNR can only be efficiently denoised with a few wavelets. Either a single wavelet or a collection of optimal wavelets (i.e., top five wavelets) should be selected from the highest μsc values. The code is available on GitHub and the signalsciencelab.com website.
|
|
|
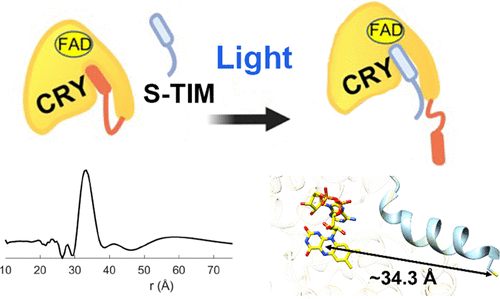
Dissecting the Interaction between Cryptochrome and Timeless Reveals Underpinnings of Light-Dependent Recognition
C. M. Schneps, R. Dunleavy, B. R. Crane
Biochemistry 63 Online ahead of print (2024)
|
|
|
Dissecting the Interaction between Cryptochrome and Timeless Reveals Underpinnings of Light-Dependent Recognition
C. M. Schneps, R. Dunleavy, B. R. Crane
Biochemistry 63 Online ahead of print (2024)
<doi: 10.1021/acs.biochem.3c00630>
PMID:
38294880
PMCID:
PMC11289166
|
|
|
ABSTRACT: Circadian rhythms are determined by cell-autonomous transcription–translation feedback loops that entrain to environmental stimuli. In the model circadian clock of Drosophila melanogaster, the clock is set by the light-induced degradation of the core oscillator protein timeless (TIM) by the principal light-sensor cryptochrome (CRY). The cryo-EM structure of CRY bound to TIM revealed that within the extensive CRY:TIM interface, the TIM N-terminus binds into the CRY FAD pocket, in which FAD and the associated phosphate-binding loop (PBL) undergo substantial rearrangement. The TIM N-terminus involved in CRY binding varies in isoforms that facilitate the adaptation of flies to different light environments. Herein, we demonstrate, through peptide binding assays and pulsed-dipolar electron spin resonance (ESR) spectroscopy, that the TIM N-terminal peptide alone exhibits light-dependent binding to CRY and that the affinity of the interaction depends on the initiating methionine residue. Extensions to the TIM N-terminus that mimic less light-sensitive variants have substantially reduced interactions with CRY. Substitutions of CRY residues that couple to the flavin rearrangement in the CRY:TIM complex have dramatic effects on CRY light activation. CRY residues Arg237 on α8, Asn253, and Gln254 on the PBL are critical for the release of the CRY autoinhibitory C-terminal tail (CTT) and subsequent TIM binding. These key light-responsive elements of CRY are well conserved throughout Type I cryptochromes of invertebrates but not by cryptochromes of chordates and plants, which likely utilize a distinct light-activation mechanism.
|
|
|
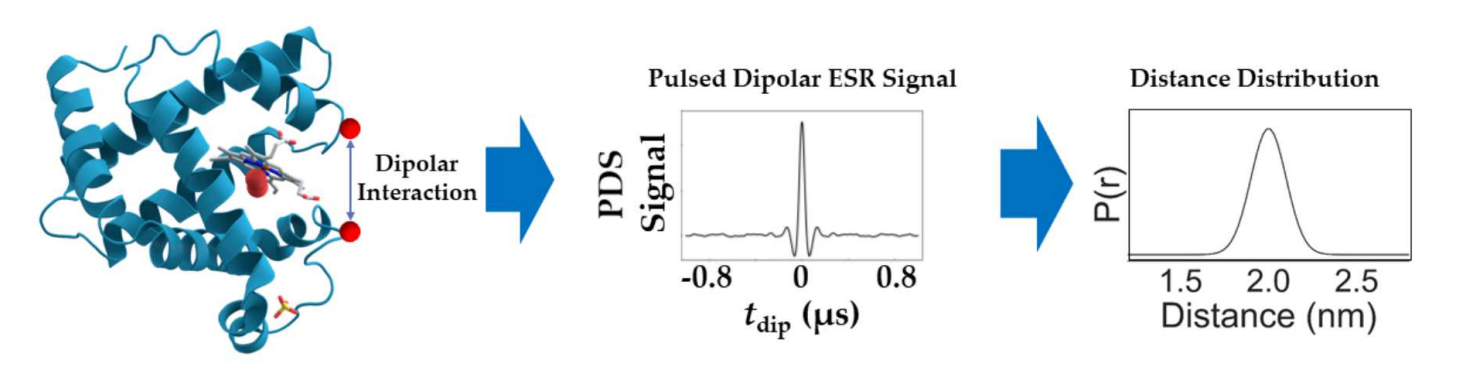
Differentiating Unimodal and Multimodal Distributions in Pulsed Dipolar Spectroscopy Using Wavelet Transforms
A.Sinha Roy, J. H. Freed, M. Srivastava
Appl. Magn. Reson. 55 (1-3) 219-237 (2024)
Supporting Information
<doi: 10.1007/s00723-023-01616-w>
PMID:
37577617
PMCID:
PMC10418556
|
|
|
ABSTRACT: Site directed spin labeling has enabled protein structure determination using electron spin resonance (ESR) pulsed dipolar spectroscopy (PDS). Small details in a distance distribution can be key to understanding important protein structure-function relationships. A major challenge has been to differentiate unimodal and overlapped multimodal distance distributions. They often yield similar distributions and dipolar signals. Current model-free distance reconstruction techniques such as Srivastava-Freed Singular Value Decomposition (SF-SVD) and Tikhonov regularization can suppress these small features in uncertainty and/or error bounds, despite being present. In this work, we demonstrate that continuous wavelet transform (CWT) can distinguish PDS signals from unimodal and multimodal distance distributions. We show that periodicity in CWT representation reflects unimodal distributions, which is masked for multimodal cases. This work is meant as a precursor to a cross-validation technique, which could indicate the modality of the distance distribution.
|
|
|
Enzymatic Spin-Labeling of Protein N- and C-Termini for Electron Paramagnetic Resonance Spectroscopy
R. Dunleavy, S. Chandrasekaran, and B. R. Crane
Bioconj. Chem. 34, Online ahead of print (2023)
Supporting Information
<doi: 10.1021/acs.bioconjchem.3c00029>
PMID:
36921260
PMCID:
PMC10502183
|

|
|
ABSTRACT: Electron paramagnetic resonance (EPR) spectroscopy is a powerful tool for investigating the structure and dynamics of proteins. The introduction of paramagnetic moieties at specific positions in a protein enables precise measurement of local structure and dynamics. This technique, termed site-directed spin-labeling, has traditionally been performed using cysteine-reactive radical-containing probes. However, large proteins are more likely to contain multiple cysteine residues and cysteine labeling at specific sites may be infeasible or impede function. To address this concern, we applied three peptide-ligating enzymes (sortase, asparaginyl endopeptidase, and inteins) for nitroxide labeling of N- and C-termini of select monomeric and dimeric proteins. Continuous wave and pulsed EPR (double electron electron resonance) experiments reveal specific attachment of nitroxide probes to ether N-termini (OaAEP1) or C-termini (sortase and intein) across three test proteins (CheY, CheA, and iLOV), thereby enabling a straightforward, highly specific, and general method for protein labeling. Importantly, the linker length (3, 5, and 9 residues for OaAEP1, intein, and sortase reactions, respectively) between the probe and the target protein has a large impact on the utility of distance measurements by pulsed EPR, with longer linkers leading to broader distributions. As these methods are only dependent on accessible N- and C-termini, we anticipate application to a wide range of protein targets for biomolecular EPR spectroscopy.
|
|
|
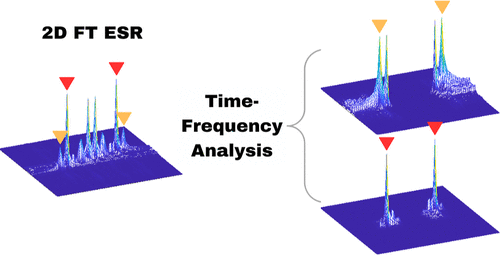
Time-Frequency Analysis of Two-Dimensional Electron Spin Resonance Signals
G. R. Sahoo, A. Sinha Roy, M. Srivastava
J. Phys. Chem. A 127, 7793-7801 (2023)
Supporting Information
<doi: 10.1021/acs.jpca.3c02708>
PMID:
37699569
PMCID:
PMC10529365
|
|
|
ABSTRACT: Two-dimensional electron spin resonance (2D ESR) spectroscopy is a unique experimental technique for probing protein structure and dynamics, including processes that occur at the microsecond time scale. While it provides significant resolution enhancement over the one-dimensional experimental setup, spectral broadening and noise make extraction of spectral information highly challenging. Traditionally, two-dimensional Fourier transform (2D FT) is applied for the analysis of 2D ESR signals, although its efficiency is limited to stationary signals. In addition, it often fails to resolve overlapping peaks in 2D ESR. In this work, we propose a time-frequency analysis of 2D time-domain signals, which identifies all frequency peaks by decoupling a signal into its distinct constituent components via projection on the time-frequency plane. The method utilizes 2D undecimated discrete wavelet transform (2D UDWT) as an intermediate step in the analysis, followed by signal reconstruction and 2D FT. We have applied the method to a simulated 2D double quantum coherence (DQC) signal for validation and a set of experimental 2D ESR signals, demonstrating its efficiency in resolving overlapping peaks in the frequency domain, while displaying frequency evolution with time in case of non-stationary data.
|
|
|
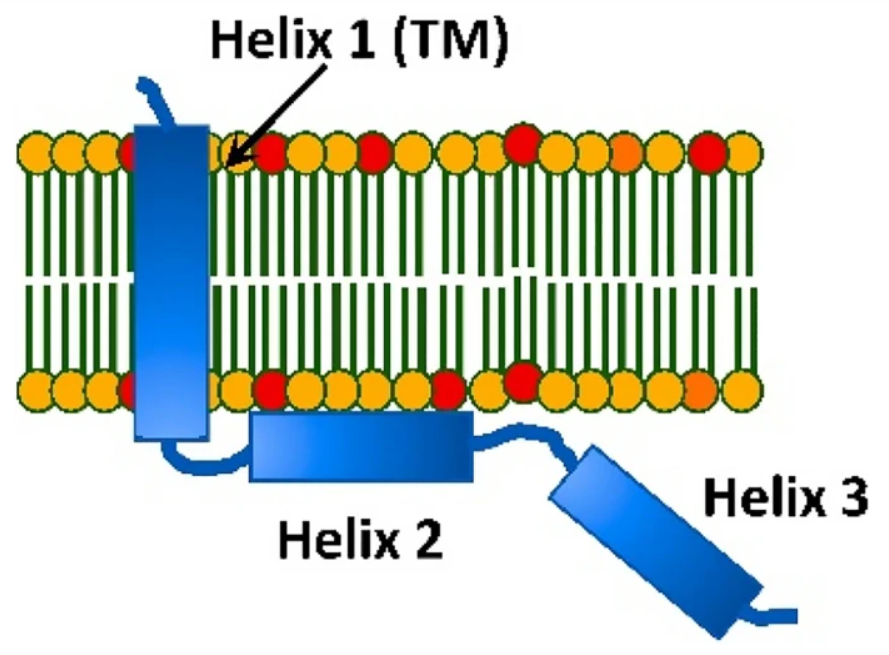
HIV-1 Vpu protein forms stable oligomers in aqueous solution via its transmembrane domain self-association
S. Majeed, L. Dang, M. M. Islam, O. Ishola, P. P. Borbat, S. J. Ludtke, and E. R. Georgieva
Scientific Reports 13, 14691 (2023)
Supporting Information
<doi: 10.1038/s41598-023-41873-0>
PMID:
37673923
PMCID:
PMC10483038
|
|
|
ABSTRACT: We report our findings on the assembly of the HIV-1 protein Vpu into soluble oligomers. Vpu is a key HIV-1 protein. It has been considered exclusively a single-pass membrane protein. Previous observations show that this protein forms stable oligomers in aqueous solution, but details about these oligomers still remain obscure. This is an interesting and rather unique observation, as the number of proteins transitioning between soluble and membrane embedded states is limited. In this study we made use of protein engineering, size exclusion chromatography, cryoEM and electron paramagnetic resonance (EPR) spectroscopy to better elucidate the nature of the soluble oligomers. We found that Vpu oligomerizes via its N-terminal transmembrane domain (TM). CryoEM suggests that the oligomeric state most likely is a hexamer/heptamer equilibrium. Both cryoEM and EPR suggest that, within the oligomer, the distal C-terminal region of Vpu is highly flexible. Our observations are consistent with both the concept of specific interactions among TM helices or the core of the oligomers being stabilized by hydrophobic forces. While this study does not resolve all of the questions about Vpu oligomers or their functional role in HIV-1 it provides new fundamental information about the size and nature of the oligomeric interactions.
|
|
|
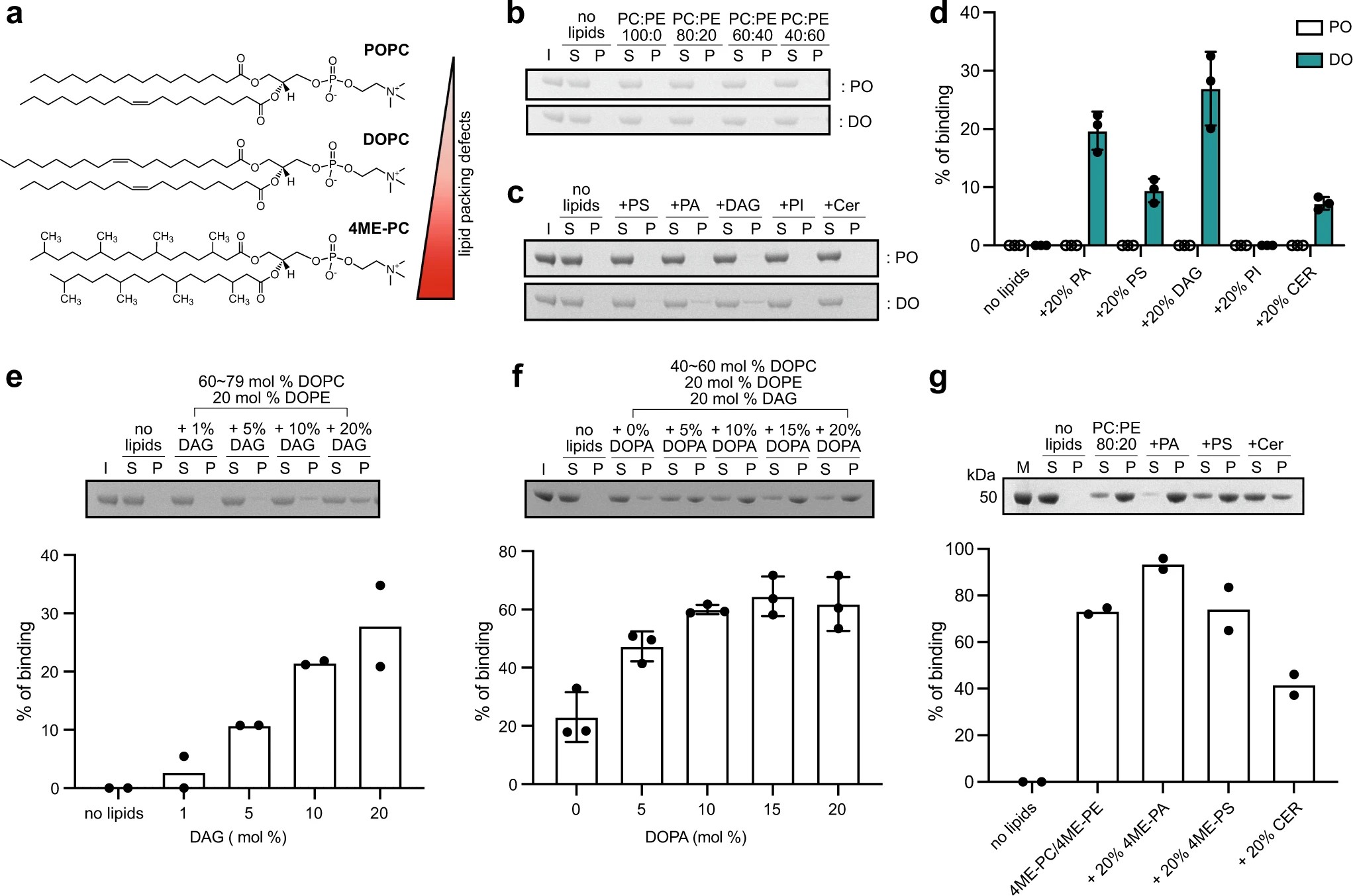
Structural insights into perilipin 3 membrane association in response to diacylglycerol accumulation
Y. M. Choi, D. Ajjaji, K. D. Fleming, P. P. Borbat, M. L. Jenkins, B. E. Moeller, S. Fernando, S. R. Bhatia, J. H. Freed, J. E. Burke, A. R. Thiam, M. V. Airola
Nat. Commun. 14 3204 (2023)
|
|
|
Structural insights into perilipin 3 membrane association in response to diacylglycerol accumulation
Y. M. Choi, D. Ajjaji, K. D. Fleming, P. P. Borbat, M. L. Jenkins, B. E. Moeller, S. Fernando, S. R. Bhatia, J. H. Freed, J. E. Burke, A. R. Thiam, M. V. Airola
Nat. Commun. 14 3204 (2023)
Supporting Information
<doi: 10.1038/s41467-023-38725-w>
PMID:
37268630
PMCID:
PMC10238389
|
|
|
ABSTRACT: Lipid droplets (LDs) are dynamic organelles that contain an oil core mainly composed of triglycerides (TAG) that is surrounded by a phospholipid monolayer and LD-associated proteins called perilipins (PLINs). During LD biogenesis, perilipin 3 (PLIN3) is recruited to nascent LDs as they emerge from the endoplasmic reticulum. Here, we analyze how lipid composition affects PLIN3 recruitment to membrane bilayers and LDs, and the structural changes that occur upon membrane binding. We find that the TAG precursors phosphatidic acid and diacylglycerol (DAG) recruit PLIN3 to membrane bilayers and define an expanded Perilipin-ADRP-Tip47 (PAT) domain that preferentially binds DAG-enriched membranes. Membrane binding induces a disorder to order transition of alpha helices within the PAT domain and 11-mer repeats, with intramolecular distance measurements consistent with the expanded PAT domain adopting a folded but dynamic structure upon membrane binding. In cells, PLIN3 is recruited to DAG-enriched ER membranes, and this requires both the PAT domain and 11-mer repeats. This provides molecular details of PLIN3 recruitment to nascent LDs and identifies a function of the PAT domain of PLIN3 in DAG binding.
|
|
|
Thermal degradation of thaumatin at low pH and its prevention using alkyl gallates
B. Pomon, Y. Zhao, A. L. Lai, T. Lin, J. H. Freed, A. Abbaspourrad
Food Hydrocoll. 139, 108544 (2023)
Supporting Information
<doi: 10.1016/j.foodhyd.2023.108544>
PMID:
37546699
PMCID:
PMC10399911
|

|
|
ABSTRACT: Thaumatin, a potent sweet tasting protein extracted from the Katemfe Plant, is emerging as a natural alternative to synthetic non-nutritive sweeteners and flavor enhancer. As a food additive, its stability within the food matrix during thermal processing is of great interest to the food industry. When heated under neutral or basic conditions, thaumatin was found to lose its sweetness due to protein aggregation caused by sulfhydryl catalyzed disulfide bond interchange. At lower pH, while thaumatin was also found to lose sweetness after heating, it does so at a slower rate and shows more resistance to sweetness loss. SDS-PAGE indicated that thaumatin fragmented into multiple smaller pieces under heating in acidic pH. Using BEMPO-3, a lipophilic spin trap, we were able to detect the presence of a free-radical within the hydrophobic region of the protein during heating. Protein carbonyl content, a byproduct of protein oxidation, also increased upon heating, providing additional evidence for protein cleavage by a radical pathway. Hexyl gallate successfully inhibited the radical generation as well as protein carbonyl formation of thaumatin during heating.
|
|
|
A Simulation Independent Analysis of Single- and Multi-Component cw ESR Spectra
A. Sinha Roy, B. Dzikovski, D. Dolui, O. Makhlynets, A. Dutta, M. Srivastava
Magnetochemistry 9, 112 (2023)
Supporting Information
<doi: 10.3390/magnetochemistry9050112>
PMID:
37476293
PMCID:
PMC10357894
|
|
|
ABSTRACT: The accurate analysis of continuous-wave electron spin resonance (cw ESR) spectra of biological or organic free-radicals and paramagnetic metal complexes is key to understanding their structure–function relationships and electrochemical properties. The current methods of analysis based on simulations often fail to extract the spectral information accurately. In addition, such analyses are highly sensitive to spectral resolution and artifacts, users' defined input parameters and spectral complexity. We introduce a simulation-independent spectral analysis approach that enables broader application of ESR. We use a wavelet packet transform-based method for extracting g values and hyperfine (A) constants directly from cw ESR spectra. We show that our method overcomes the challenges associated with simulation-based methods for analyzing poorly/partially resolved and unresolved spectra, which is common in most cases. The accuracy and consistency of the method are demonstrated on a series of experimental spectra of organic radicals and copper–nitrogen complexes. We showed that for a two-component system, the method identifies their individual spectral features even at a relative concentration of 5% for the minor component.
|
|
|
Insights into the oligomeric structure of the HIV-1 Vpu protein
S. Majeed, O. Adetuyi, P. P. Borbat, M. M. Islam, O. Ishola, B. Zhao, E. R. Georgieva
J. Struct. Biol. 215, 107943 (2023)
Supporting Information
<doi: 10.1016/j.jsb.2023.107943>
PMID:
36796461
PMCID:
PMC10257199
|
|
|
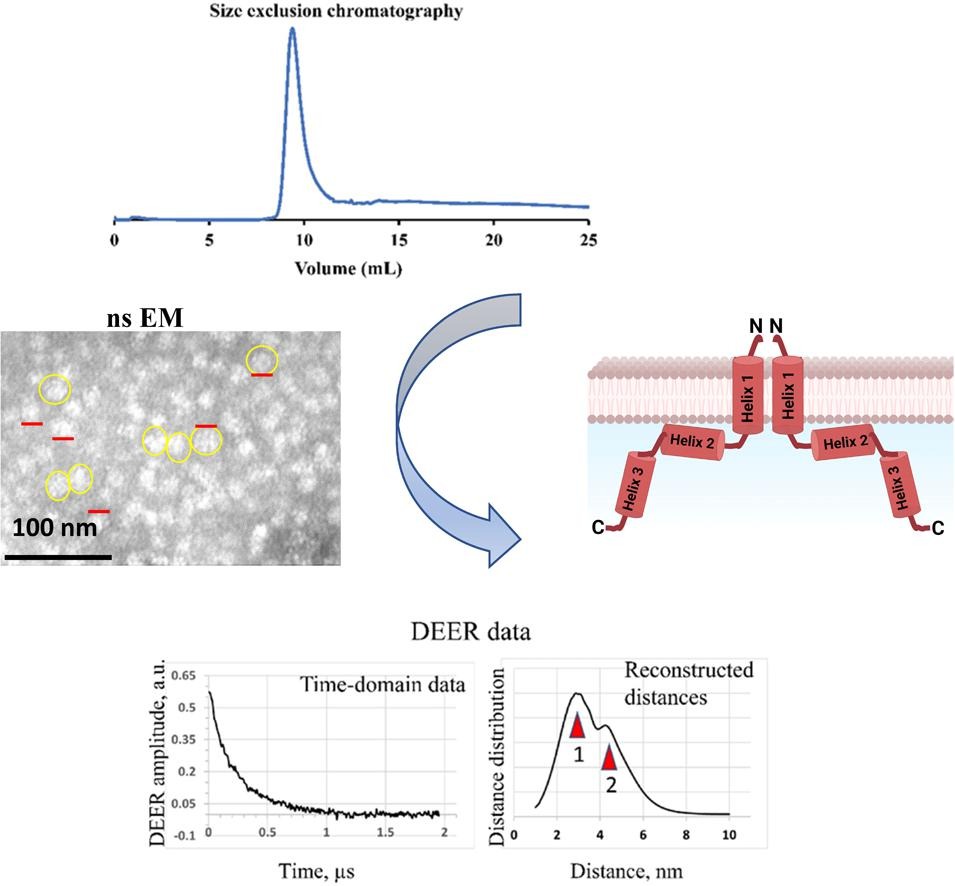
ABSTRACT: The HIV-1-encoded protein Vpu forms an oligomeric ion channel/pore in membranes and interacts with host proteins to support the virus lifecycle. However, Vpu molecular mechanisms are currently not well understood. Here, we report on the Vpu oligomeric organization under membrane and aqueous conditions and provide insights into how the Vpu environment affects the oligomer formation. For these studies, we designed a maltose-binding protein (MBP)-Vpu chimera protein and produced it in E. coli in soluble form. We analyzed this protein using analytical size-exclusion chromatography (SEC), negative staining electron microscopy (nsEM), and electron paramagnetic resonance (EPR) spectroscopy. Surprisingly, we found that MBP-Vpu formed stable oligomers in solution, seemingly driven by Vpu transmembrane domain self-association. A coarse modeling of nsEM data as well as SEC and EPR data suggests that these oligomers most likely are pentamers, similar to what was reported regarding membrane-bound Vpu. We also noticed reduced MBP-Vpu oligomer stability upon reconstitution of the protein in β-DDM detergent and mixtures of lyso-PC/PG or DHPC/DHPG. In these cases, we observed greater oligomer heterogeneity, with MBP-Vpu oligomeric order generally lower than in solution; however, larger oligomers were also present. Notably, we found that in lyso-PC/PG, above a certain protein concentration, MBP-Vpu assembles into extended structures, which had not been reported for Vpu. Therefore, we captured various Vpu oligomeric forms, which can shed light on Vpu quaternary organization. Our findings could be useful in understanding Vpu organization and function in cellular membranes and could provide information regarding the biophysical properties of single-pass transmembrane proteins.
|
|
|
Membrane Binding Induces Distinct Structural Signatures in the Mouse Complexin–1C–Terminal Domain
E. M. Grasso, M. S. Terakawa, A. L. Lai, Y. X. Xie, T. F. Ramlall, J. H. Freed, and D. Eliezer.
J. Mol. Biol. 435, 167710 (2023)
Supporting Information
<doi: 10.1016/j.jmb.2022.167710>
PMID:
35777466
PMCID:
PMC9794636
|
|
|
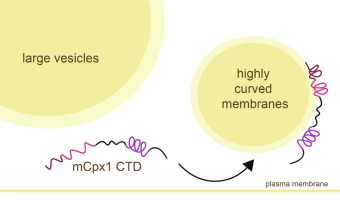
ABSTRACT: Complexins play a critical role in regulating SNARE-mediated exocytosis of synaptic vesicles. Evolutionary divergences in complexin function have complicated our understanding of the role these proteins play in inhibiting the spontaneous fusion of vesicles. Previous structural and functional characterizations of worm and mouse complexins have indicated the membrane curvature-sensing C-terminal domain of these proteins is responsible for differences in inhibitory function. We have characterized the structure and dynamics of the mCpx1 CTD in the absence and presence of membranes and membrane mimetics using NMR, ESR, and optical spectroscopies. In the absence of lipids, the mCpx1 CTD features a short helix near its N-terminus and is otherwise disordered. In the presence of micelles and small unilamellar vesicles, the mCpx1 CTD forms a discontinuous helical structure in its C-terminal 20 amino acids, with no preference for specific lipid compositions. In contrast, the mCpx1 CTD shows distinct compositional preferences in its interactions with large unilamellar vesicles. These studies identify structural divergences in the mCpx1 CTD relative to the wCpx1 CTD in regions that are known to be critical to the wCpx1 CTD's role in inhibiting spontaneous fusion of synaptic vesicles, suggesting a potential structural basis for evolutionary divergences in complexin function.
|
|
|
Unsupervised Analysis of Small Molecule Mixtures by Wavelet-Based Super-Resolved NMR
A. Sinha Roy, M. Srivastava
Molecules 28 792 (2023)
Supporting Information
<doi: 10.3390/molecules28020792>
PMID:
36677850
PMCID:
PMC9866129
|
|
|
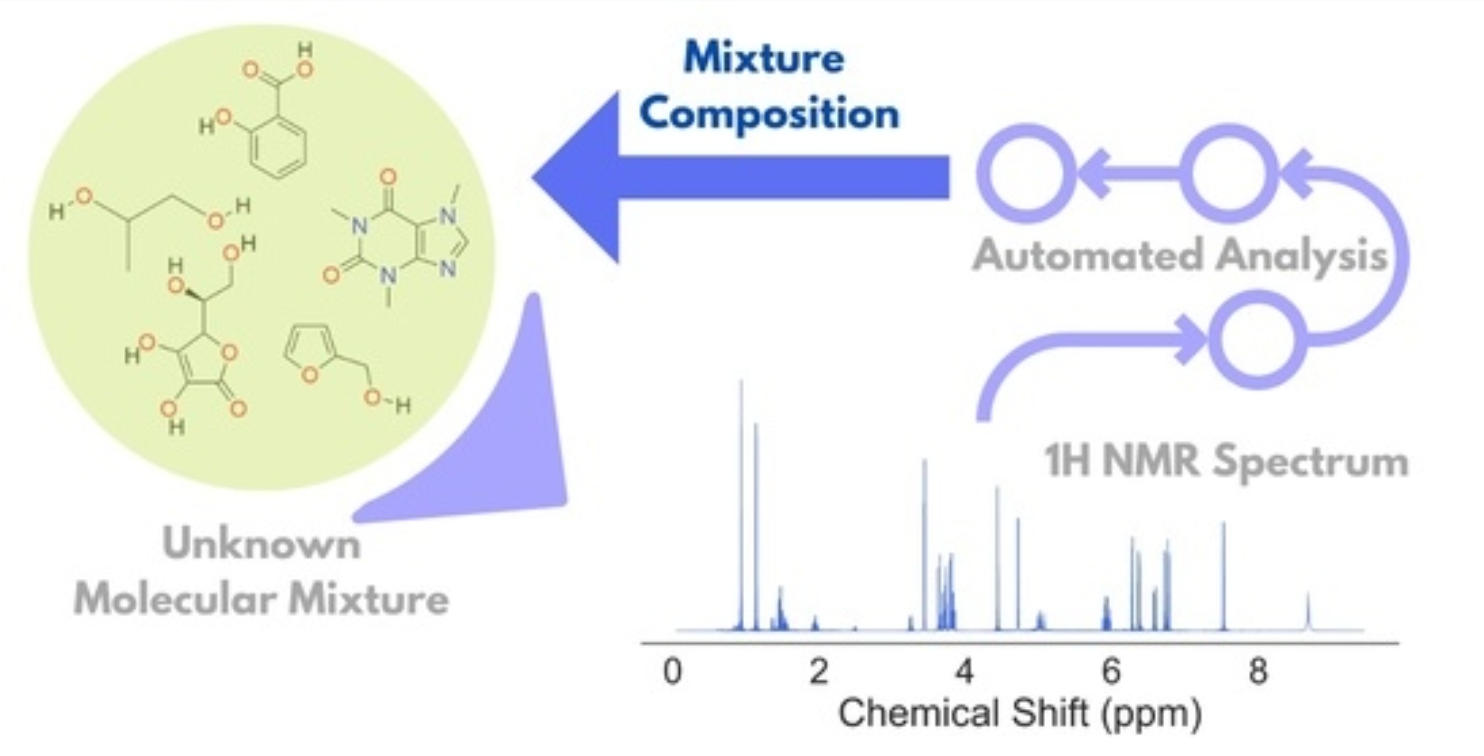
ABSTRACT: Resolving small molecule mixtures by nuclear magnetic resonance (NMR) spectroscopy has been of great interest for a long time for its precision, reproducibility, and efficiency. However, spectral analyses for such mixtures are often highly challenging due to overlapping resonance lines and limited chemical shift windows. The existing experimental and theoretical methods to produce shift NMR spectra in dealing with the problem have limited applicability owing to sensitivity issues, inconsistency, and/or the requirement of prior knowledge. Recently, we resolved the problem by decoupling multiplet structures in NMR spectra by the wavelet packet transform (WPT) technique. In this work, we developed a scheme for deploying the method in generating highly resolved WPT NMR spectra and predicting the composition of the corresponding molecular mixtures from their 1H NMR spectra in an automated fashion. The four-step spectral analysis scheme consists of calculating the WPT spectrum, peak matching with a WPT shift NMR library, followed by two optimization steps in producing the predicted molecular composition of a mixture. The robustness of the method was tested on an augmented dataset of 1000 molecular mixtures, each containing 3 to 7 molecules. The method successfully predicted the constituent molecules with a median true positive rate of 1.0 against the varying compositions, while a median false positive rate of 0.04 was obtained. The approach can be scaled easily for much larger datasets.
|
|
|
A Non-Perturbative, Low-Noise Surface Coating for Sensitive Force-Gradient Detection of Electron Spin Resonance in Thin Films
M. C. Boucher, C. E. Isaac, P. Sun, P. P. Borbat, and J. A. Marohn
ACS Nano 17, 2c08635 (2023)
Supporting Information
<doi: 10.1021/acsnano.2c08635>
PMID:
36625878
PMCID:
PMC10330945
|
|
|
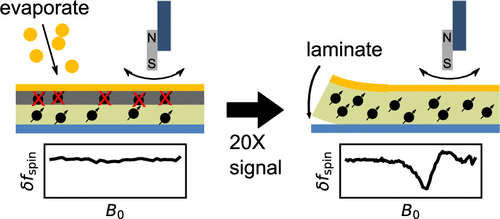
ABSTRACT: The sensitivity of magnetic resonance force microscopy (MRFM) is limited by surface noise. Coating a thin-film polymer sample with metal has been shown to decrease, by orders of magnitude, sample-related force noise and frequency noise in MRFM experiments. Using both MRFM and inductively detected measurements of electron-spin resonance, we show that thermally evaporating a 12 nm gold layer on a 40 nm nitroxide-doped polystyrene film inactivates the nitroxide spin labels to a depth of 20 nm, making single-spin measurements difficult or impossible. We introduce a "laminated sample" protocol in which the gold layer is first evaporated on a sacrificial polymer. The sample is deposited on the room-temperature gold layer, removed using solvent lift-off, and placed manually on a coplanar waveguide. Electron spin resonance (ESR) of such a laminated sample was detected via MRFM at cryogenic temperatures using a high-compliance cantilever with an integrated 100-nm-scale cobalt tip. A 20-fold increase of spin signal was observed relative to a thin-film sample prepared instead with an evaporated metal coating. The observed signal is still somewhat smaller than expected, and we discuss possible remaining sources of signal loss.
|
|
|
Analysis of Small-Molecule Mixtures by Super-Resolved 1H NMR Spectroscopy
A. Sinha Roy and M. Srivastava
J. Phys. Chem. A 126, 9108–9113 (2022)
Supporting Information
<doi: 10.1021/acs.jpca.2c06858>
PMID:
36413171
PMCID:
PMC10228708
|

|
|
ABSTRACT: Analysis of small molecules is essential to metabolomics, natural products, drug discovery, food technology, and many other areas of interest. Current barriers preclude from identifying the constituent molecules in a mixture as overlapping clusters of NMR lines pose a major challenge in resolving signature frequencies for individual molecules. While homonuclear decoupling techniques produce much simplified pure shift spectra, they often affect sensitivity. Conversion of typical NMR spectra to pure shift spectra by signal processing without a priori knowledge about the coupling patterns is essential for accurate analysis. We developed a super-resolved wavelet packet transform based 1H NMR spectroscopy that can be used in high-throughput studies to reliably decouple individual constituents of small molecule mixtures. We demonstrate the efficacy of the method on the model mixtures of saccharides and amino acids in the presence of significant noise.
|
|
|
Interdomain Linkers Regulate Histidine Kinase Activity by Controlling Subunit Interactions
Z. Maschmann, S. Chandrasekaran, T. K. Chua, B. R. Crane
Biochemistry 61, 2672-2686 (2022)
Supporting Information
<doi: 10.1021/acs.biochem.2c00326>
PMID:
36321948
PMCID:
PMC10134573
|
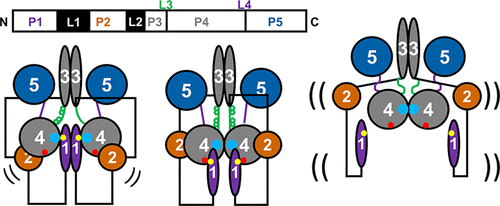
|
|
ABSTRACT: Bacterial chemoreceptors regulate the cytosolic multidomain histidine kinase CheA through largely unknown mechanisms. Residue substitutions in the peptide linkers that connect the P4 kinase domain to the P3 dimerization and P5 regulatory domain affect CheA basal activity and activation. To understand the role that these linkers play in CheA activity, the P3-to-P4 linker (L3) and P4-to-P5 linker (L4) were extended and altered in variants of Thermotoga maritima (Tm) CheA. Flexible extensions of the L3 and L4 linkers in CheA-LV1 (linker variant 1) allowed for a well-folded kinase domain that retained wild-type (WT)-like binding affinities for nucleotide and normal interactions with the receptor-coupling protein CheW. However, CheA-LV1 autophosphorylation activity registered ~50-fold lower compared to WT. Neither a WT nor LV1 dimer containing a single P4 domain could autophosphorylate the P1 substrate domain. Autophosphorylation activity was rescued in variants with extended L3 and L4 linkers that favor helical structure and heptad spacing. Autophosphorylation depended on linker spacing and flexibility and not on sequence. Pulse-dipolar electron-spin resonance (ESR) measurements with spin-labeled adenosine 5′-triphosphate (ATP) analogues indicated that CheA autophosphorylation activity inversely correlated with the proximity of the P4 domains within the dimers of the variants. Despite their separation in primary sequence and space, the L3 and L4 linkers also influence the mobility of the P1 substrate domains. In all, interactions of the P4 domains, as modulated by the L3 and L4 linkers, affect domain dynamics and autophosphorylation of CheA, thereby providing potential mechanisms for receptors to regulate the kinase.
|
|
|
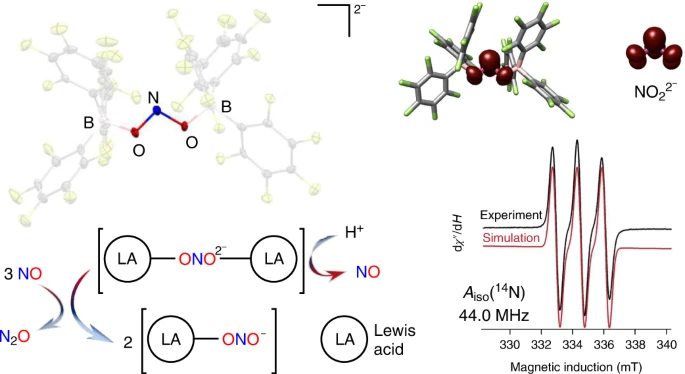
Lewis acid-assisted reduction of nitrite to nitric and nitrous oxides via the elusive nitrite radical dianion
V. Hosseininasab, I. M DiMucci, P. Ghosh, J. A. Bertke, S. Chandrasekharan, C. J. Titus, D. Nordlund, J. H. Freed, K. M. Lancaster, T. H. Warren
Nat. Chem. 14, 1265-1269 (2022)
|
|
|
Lewis acid-assisted reduction of nitrite to nitric and nitrous oxides via the elusive nitrite radical dianion
V. Hosseininasab, I. M DiMucci, P. Ghosh, J. A. Bertke, S. Chandrasekharan, C. J. Titus, D. Nordlund, J. H. Freed, K. M. Lancaster, T. H. Warren
Nat. Chem. 14, 1265-1269 (2022)
Supporting Information
<doi: 10.1038/s41557-022-01025-9>
PMID:
36064970
PMCID:
PMC9633411
|
|
|
ABSTRACT: Reduction of nitrite anions (NO2–) to nitric oxide (NO), nitrous oxide (N2O) and ultimately dinitrogen (N2) takes place in a variety of environments, including in the soil as part of the biogeochemical nitrogen cycle and in acidified nuclear waste. Nitrite reduction typically takes place within the coordination sphere of a redox-active transition metal. Here we show that Lewis acid coordination can substantially modify the reduction potential of this polyoxoanion to allow for its reduction under non-aqueous conditions (–0.74 V versus NHE). Detailed characterization confirms the formation of the borane-capped radical nitrite dianion (NO22–), which features a N(II) oxidation state. Protonation of the nitrite dianion results in the facile loss of nitric oxide (NO), whereas its reaction with NO results in disproportionation to nitrous oxide (N2O) and nitrite (NO2–). This system connects three redox levels in the global nitrogen cycle and provides fundamental insights into the conversion of NO2– to NO.
|
|
|
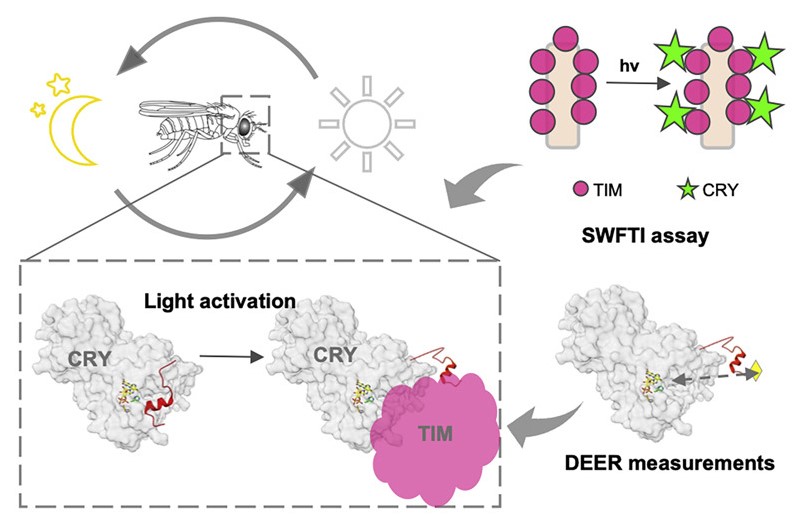
Mechanistic insight into light-dependent recognition of Timeless by Drosophila Cryptochrome
C. Lin, C. M. Schneps, S. Chandrasekaran, A. Ganguly, B. R. Crane
Structure 30, 851-861 (2022)
|
|
|
Mechanistic insight into light-dependent recognition of Timeless by Drosophila Cryptochrome
C. Lin, C. M. Schneps, S. Chandrasekaran, A. Ganguly, B. R. Crane
Structure 30, 851-861 (2022)
<doi: 10.1016/j.str.2022.03.010>
PMID:
35397203
PMCID:
PMC9201872
|
|
|
ABSTRACT: Cryptochrome (CRY) entrains the fly circadian clock by binding to Timeless (TIM) in light. Undocking of a helical C-terminal tail (CTT) in response to photoreduction of the CRY flavin cofactor gates TIM recognition. We present a generally applicable select western-blot-free tagged-protein interaction (SWFTI) assay that allowed the quantification of CRY binding to TIM in dark and light. The assay was used to study CRY variants with residue substitutions in the flavin pocket and correlate their TIM affinities with CTT undocking, as measured by pulse-dipolar ESR spectroscopy and evaluated by molecular dynamics simulations. CRY variants with the CTT removed or undocked bound TIM constitutively, whereas those incapable of photoreduction bound TIM weakly. In response to the flavin redox state, two conserved histidine residues contributed to a robust on/off switch by mediating CTT interactions with the flavin pocket and TIM. Our approach provides an expeditious means to quantify the interactions of difficult-to-produce proteins.
|
|
|
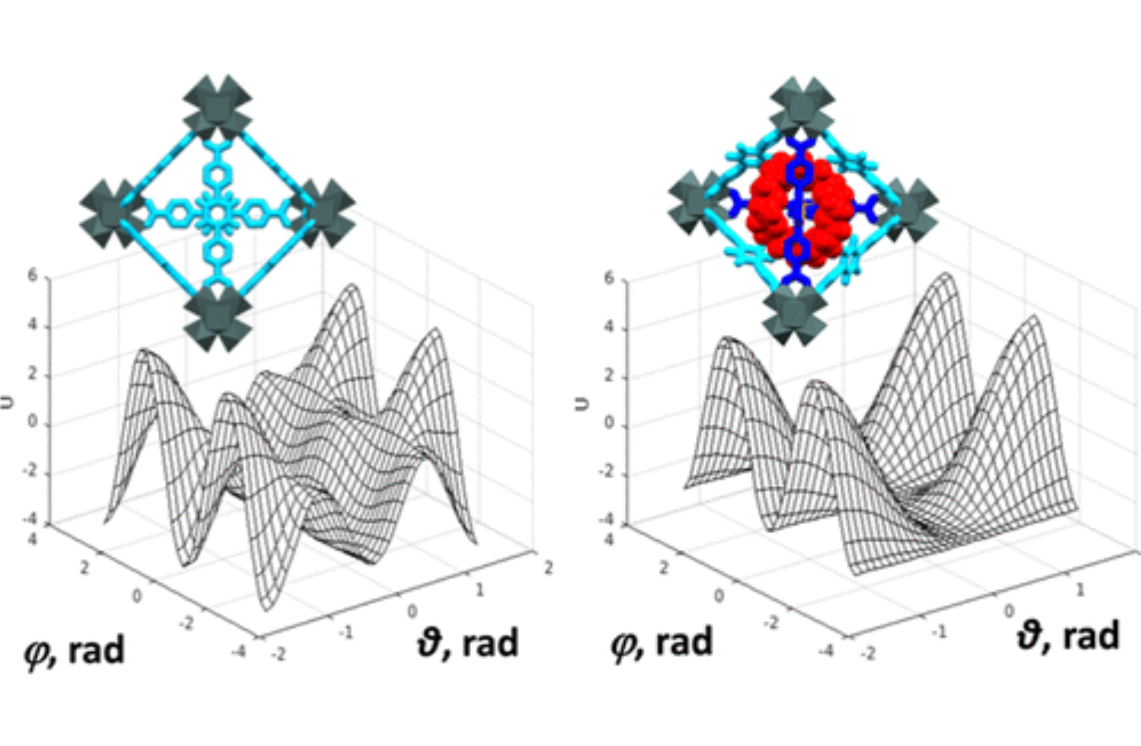
Structural Dynamics by NMR in the Solid State: II. The MOMD Perspective of the Dynamic Structure of Metal-Organic Frameworks Comprising Several Mobile Components
E. Meirovitch, Z. Liang, R. W. Schurko, S. J. Loeb, and J. H. Freed.
J. Phys. Chem. B 126, 2452-2465 (2022)
Supporting Information
<doi: 10.1021/acs.jpcb.1c10120>
PMID:
35333061
PMCID:
PMC9055879
|
|
|
ABSTRACT: We describe the application of the microscopic-order-macroscopic-disorder (MOMD) approach, developed for the analysis of dynamic 2H NMR lineshapes in the solid state, to unravel interactions among the constituents of metal–organic frameworks (MOFs) that comprise mobile components. MOMD was applied recently to University of Windsor Dynamic Material (UWDM) MOFs with one mobile crown ether per cavity. In this work, we study UWDM-9-d4, which comprises a mobile 2H-labeled phenyl-ring residue along with an isotopically unlabeled 24C8 crown ether. We also study UiO-68-d4, which is structurally similar to UWDM-9-d4 but lacks the crown ether. The physical picture consists of the NMR probe–the C–D bonds of the phenyl-d4 rotor–diffusing locally (diffusion tensor R) in the presence of a local ordering potential, u. For UiO-68-d4, we find it sufficient to expand u in terms of four real Wigner functions, D0|K|L, overall 2–3 kT in magnitude, with R∥ relatively fast, and R⊥; in the (2.8–5.0) × 102 s-1 range. For UWDM-9-d4, u requires only two terms 2–3 kT in magnitude and slower rate constants R∥ and R⊥. In the more crowded macrocycle-containing UWDM-9-d4 cavity, phenyl-d4 dynamics is more isotropic and is described by a simpler ordering potential. This is ascribed to cooperative phenyl-ring/macrocycle motion, which yields a dynamic structure more uniform in character. The experimental 2H spectra used here were analyzed previously with a multi-simple-mode (MSM) approach where several independent simple motional modes are combined. Where possible, similar features have been identified and used to compare the two approaches.
|
|
|
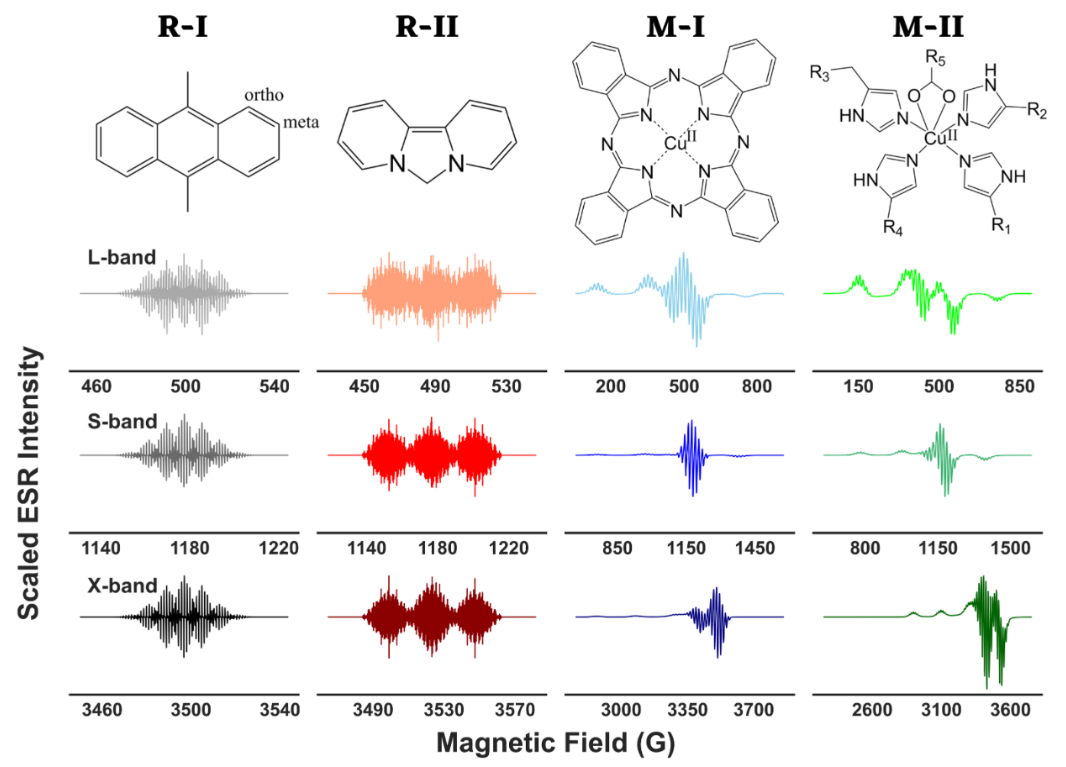
Hyperfine Decoupling of ESR Spectra Using Wavelet Transform
A. Sinha Roy and M. Srivastava
Magnetochemistry 8, 32 (2022)
<doi: 10.3390/magnetochemistry8030032>
PMID:
37475982
PMCID:
PMC10357921
|
|
|
ABSTRACT: The objective of spectral analysis is to resolve and extract relevant features from experimental data in an optimal fashion. In continuous-wave (cw) electron spin resonance (ESR) spectroscopy, both g values of a paramagnetic center and hyperfine splitting (A) caused by its interaction with neighboring magnetic nuclei in a molecule provide important structural and electronic information. However, in the presence of g- and/or A-anisotropy and/or large number of resonance lines, spectral analysis becomes highly challenging. Either high-resolution experimental techniques are employed to resolve the spectra in those cases or a range of suitable ESR frequencies are used in combination with simulations to identify the corresponding g and A values. In this work, we present a wavelet transform technique in resolving both simulated and experimental cw-ESR spectra by separating the hyperfine and super-hyperfine components. We exploit the multiresolution property of wavelet transforms that allow the separation of distinct features of a spectrum based on simultaneous analysis of spectrum and its varying frequency. We retain the wavelet components that stored the hyperfine and/or super-hyperfine features, while eliminating the wavelet components representing the remaining spectrum. We tested the method on simulated cases of metal–ligand adducts at L-, S-, and X-band frequencies, and showed that extracted g values, hyperfine and super-hyperfine coupling constants from simulated spectra, were in excellent agreement with the values of those parameters used in the simulations. For the experimental case of a copper(II) complex with distorted octahedral geometry, the method was able to extract g and hyperfine coupling constant values, and revealed features that were buried in the overlapped spectra.
|
|
|
Revisiting signal analysis in the big data era
M. Srivastava
Nature Computational Science 2, 70-71 (2022)
<doi: 10.1038/s43588-022-00210-7>
PMID:
37274540
PMCID:
PMC10238072
|

|
|
ABSTRACT: A fast and accurate time–frequency analysis is challenging for many applications, especially in the current big data era. A recent work introduces a fast continuous wavelet transform that effectively boosts the analysis speed without sacrificing the resolution of the result.
|
|
|
The N-Terminal Domain of Aβ40-Amyloid Fibril: The MOMD Perspective of its Dynamic Structure from NMR Lineshape Analysis
E. Meirovitch, Z. Liang, and J. H. Freed.
J. Phys. Chem. B 126, 1202-1211 (2022)
Supporting Information
<doi: 10.1021/acs.jpcb.1c10131>
PMID:
35128920
PMCID:
PMC8908910
|
|
|
ABSTRACT: We have developed the stochastic microscopic‐order‐macroscopic‐disorder (MOMD) approach for elucidating dynamic structures in the solid‐state from 2H NMR lineshapes. In MOMD, the probe experiences an effective/collective motional mode. The latter is described by a potential, u, which represents the local spatial‐restrictions, a local‐motional diffusion tensor, R, and key features of local geometry. Previously we applied MOMD to the well‐structured core domain of the 3‐fold‐symmetric twisted polymorph of the Aβ40‐amyloid fibril. Here, we apply it to the N‐terminal domain of this fibril. We find that the dynamic structures of the two domains are largely similar but differ in the magnitude and complexity of the key physical parameters. This interpretation differs from previous multisimple‐mode (MSM) interpretations of the same experimental data. MSM used for the two domains different combinations of simple motional modes taken to be independent. For the core domain, MOMD and MSM disagree on the character of the dynamic structure. For the N‐terminal domain, they even disagree on whether this chain segment is structurally ordered (MOMD finds that it is), and whether it undergoes a phase transition at 260 K where bulklike water located in the fibril matrix freezes (MOMD finds that it does not). These are major differences associated with an important system. While the MOMD description is a physically sound one, there are drawbacks in the MSM descriptions. The results obtained in this study promote our understanding of the dynamic structure of protein aggregates. Thus, they contribute to the effort to pharmacologically control neurodegenerative disorders believed to be caused by such aggregates.
|
|
|
Negatively charged residues in the membrane ordering activity of SARS‐CoV‐1 and ‐2 fusion peptides
A.L. Lai and J.H. Freed
Biophys. J. 121, 207-227 (2022)
Supporting Information
<doi: 10.1016/j.bpj.2021.12.024>
PMID:
34929193
PMCID:
PMC8683214
|
|
|
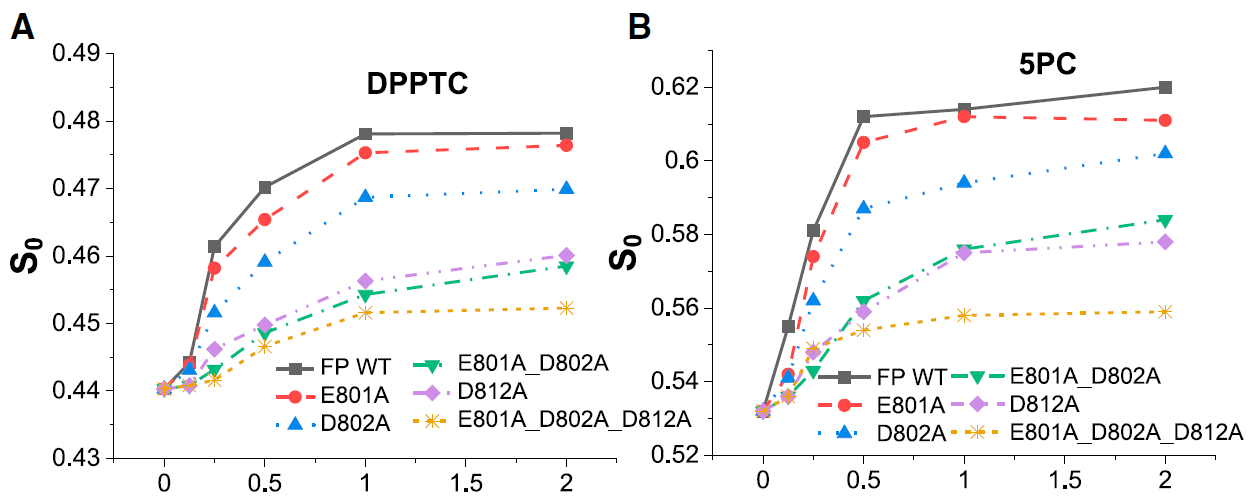
ABSTRACT: Entry of coronaviruses into host cells is mediated by the viral spike protein. Previously, we identified the bona fide fusion peptides (FPs) for severe acute respiratory syndrome coronavirus ("SARS‐1") and severe acute respiratory syndrome coronavirus‐2 ("SARS‐2") using electron spin resonance spectroscopy. We also found that their FPs induce membrane ordering in a Ca2+‐dependent fashion. Here we study which negatively charged residues in SARS‐1 FP are involved in this binding, to build a topological model and clarify the role of Ca2+. Our systematic mutation study on the SARS‐1 FP shows that all six negatively charged residues contribute to the FP's membrane ordering activity, with D812 the dominant residue. The corresponding SARS‐2 residue D830 plays an equivalent role. We provide a topological model of how the FP binds Ca2+ ions: its two segments FP1 and FP2 each bind one Ca2+. The binding of Ca2+, the folding of FP (both studied by isothermal titration calorimetry experiments), and the ordering activity correlate very well across the mutants, suggesting that the Ca2+ helps the folding of FP in membranes to enhance the ordering activity. Using a novel pseudotyped viral particle‐liposome methodology, we monitored the membrane ordering induced by the FPs in the whole spike protein in its trimer form in real time. We found that the SARS‐1 and SARS‐2 pseudotyped viral particles also induce membrane ordering to the extent that separate FPs do, and mutations of the negatively charged residues also significantly suppress the membrane ordering activity. However, the slower kinetics of the FP ordering activity versus that of the pseudotyped viral particle suggest the need for initial trimerization of the FPs.
|
|
|
Benchmark Test and Guidelines for DEER/PELDOR Experiments on Nitroxide-Labeled Biomolecules
O. Schiemann, C. A. Heubach, D. Abdullin, K. Ackermann, M. Azarkh, E. G. Bagryanskaya, M. Drescher, B. Endeward, J. H. Freed, L. Galazzo, D. Goldfarb, T. Hett, L. Esteban Hofer, L. Fábregas Ibáñez, E. J. Hustedt, S. Kucher, I. Kuprov, J. E. Lovett, A. Meyer, S. Ruthstein, S. Saxena, S. Stoll, C. R. Timmel, M. Di Valentin, H. S. Mchaourab, T. F. Prisner, B. E. Bode, E. Bordignon, M. Bennati, and G. Jeschke.
J. Am. Chem. Soc. 143, 17875-17890 (2021)
|
|
|
Benchmark Test and Guidelines for DEER/PELDOR Experiments on Nitroxide-Labeled Biomolecules
O. Schiemann, C. A. Heubach, D. Abdullin, K. Ackermann, M. Azarkh, E. G. Bagryanskaya, M. Drescher, B. Endeward, J. H. Freed, L. Galazzo, D. Goldfarb, T. Hett, L. Esteban Hofer, L. Fábregas Ibáñez, E. J. Hustedt, S. Kucher, I. Kuprov, J. E. Lovett, A. Meyer, S. Ruthstein, S. Saxena, S. Stoll, C. R. Timmel, M. Di Valentin, H. S. Mchaourab, T. F. Prisner, B. E. Bode, E. Bordignon, M. Bennati, and G. Jeschke.
J. Am. Chem. Soc. 143, 17875-17890 (2021)
Supporting Information
<doi: 10.1021/jacs.1c07371>
PMID:
34664948
PMCID:
PMC11253894
|
|
|
ABSTRACT: Distance distribution information obtained by pulsed dipolar EPR spectroscopy provides an important contribution to many studies in structural biology. Increasingly, such information is used in integrative structural modeling, where it delivers unique restraints on the width of conformational ensembles. In order to ensure reliability of the structural models and of biological conclusions, we herein define quality standards for sample preparation and characterization, for measurements of distributed dipole–dipole couplings between paramagnetic labels, for conversion of the primary time‐domain data into distance distributions, for interpreting these distributions, and for reporting results. These guidelines are substantiated by a multi‐laboratory benchmark study and by analysis of data sets with known distance distribution ground truth. The study and the guidelines focus on proteins labeled with nitroxides and on double electron–electron resonance (DEER aka PELDOR) measurements and provide suggestions on how to proceed analogously in other cases.
|
|
|
Highly Basic Clusters in the Herpes Simplex Virus 1 Nuclear Egress Complex Drive Membrane Budding by Inducing Lipid Ordering
M. K. Thorsen, A. Lai, D. P. Hoogerheide, G. C. L. Wong, J. H. Freed, and E. E. Heldwein.
mBio 12 e0154821 (2021)
Supporting Information
<doi: 10.1128/mBio.01548-21>
PMID:
34425706
PMCID:
PMC8406295
|
|
|
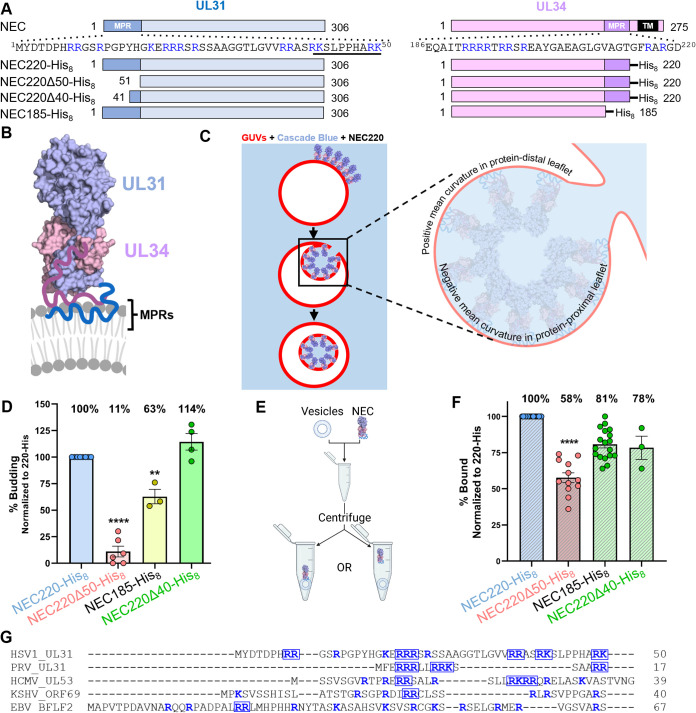
ABSTRACT: During replication of herpesviruses, capsids escape from the nucleus into the cytoplasm by budding at the inner nuclear membrane. This unusual process is mediated by the viral nuclear egress complex (NEC) that deforms the membrane around the capsid by oligomerizing into a hexagonal, membrane‐bound scaffold. Here, we found that highly basic membrane‐proximal regions (MPRs) of the NEC alter lipid order by inserting into the lipid headgroups and promote negative Gaussian curvature. We also find that the electrostatic interactions between the MPRs and the membranes are essential for membrane deformation. One of the MPRs is phosphorylated by a viral kinase during infection, and the corresponding phosphomimicking mutations block capsid nuclear egress. We show that the same phosphomimicking mutations disrupt the NEC‐membrane interactions and inhibit NEC‐mediated budding in vitro, providing a biophysical explanation for the in vivo phenomenon. Our data suggest that the NEC generates negative membrane curvature by both lipid ordering and protein scaffolding and that phosphorylation acts as an off switch that inhibits the membrane‐budding activity of the NEC to prevent capsid‐less budding.
IMPORTANCE Herpesviruses are large viruses that infect nearly all vertebrates and some invertebrates and cause lifelong infections in most of the world's population. During replication, herpesviruses export their capsids from the nucleus into the cytoplasm by an unusual mechanism in which the viral nuclear egress complex (NEC) deforms the nuclear membrane around the capsid. However, how membrane deformation is achieved is unclear. Here, we show that the NEC from herpes simplex virus 1, a prototypical herpesvirus, uses clusters of positive charges to bind membranes and order membrane lipids. Reducing the positive charge or introducing negative charges weakens the membrane deforming ability of the NEC. We propose that the virus employs electrostatics to deform nuclear membrane around the capsid and can control this process by changing the NEC charge through phosphorylation. Blocking NEC-membrane interactions could be exploited as a therapeutic strategy.
|
|
|
Determining Decomposition Levels for Wavelet Denoising Using Sparsity Plot
W. Bekerman and M. Srivastava
IEEE Access 9, 110582-110591 (2021)
|

|
|
Determining Decomposition Levels for Wavelet Denoising Using Sparsity Plot
W. Bekerman and M. Srivastava
IEEE Access 9, 110582-110591 (2021)
<doi: 10.1109/ACCESS.2021.3103497>
|
|
ABSTRACT: We present a method to select decomposition levels for noise thresholding in wavelet denoising. It is essential to determine the accurate decomposition levels to avoid inadequate noise reduction and/or signal distortion by noise thresholding. We introduce the concept of sparsity plot that captures the abrupt transition from noisy to noise-free Detail component, readily revealing the cut-off for the maximum decomposition levels. The method uses the sparsity parameter to determine the noise presence in each detail component and measures the magnitude change in the sparsity values to distinguish between noisy and noise-free Detail components. The method is tested on both model and experimental signals, and proves effective for various signal lengths and types, as well as different Signal-to-Noise Ratios (SNRs). The method can be embedded with any wavelet denoising method to improve its performance. The code is available via GitHub and denoising.cornell.edu, as well as the corresponding author’s group website ( signalsciencelab.com). |
|
|
Theory and Least Squares Fitting of CW ESR Saturation Spectra Using the MOMD Model
P. Gupta, B. Dzikovski, and J. H. Freed
Appl. Magn. Reson. 53 699-715 (2021)
<doi: 10.1007/s00723-021-01390-7>
PMID:
35431460
PMCID:
PMC9012167
|
|
|
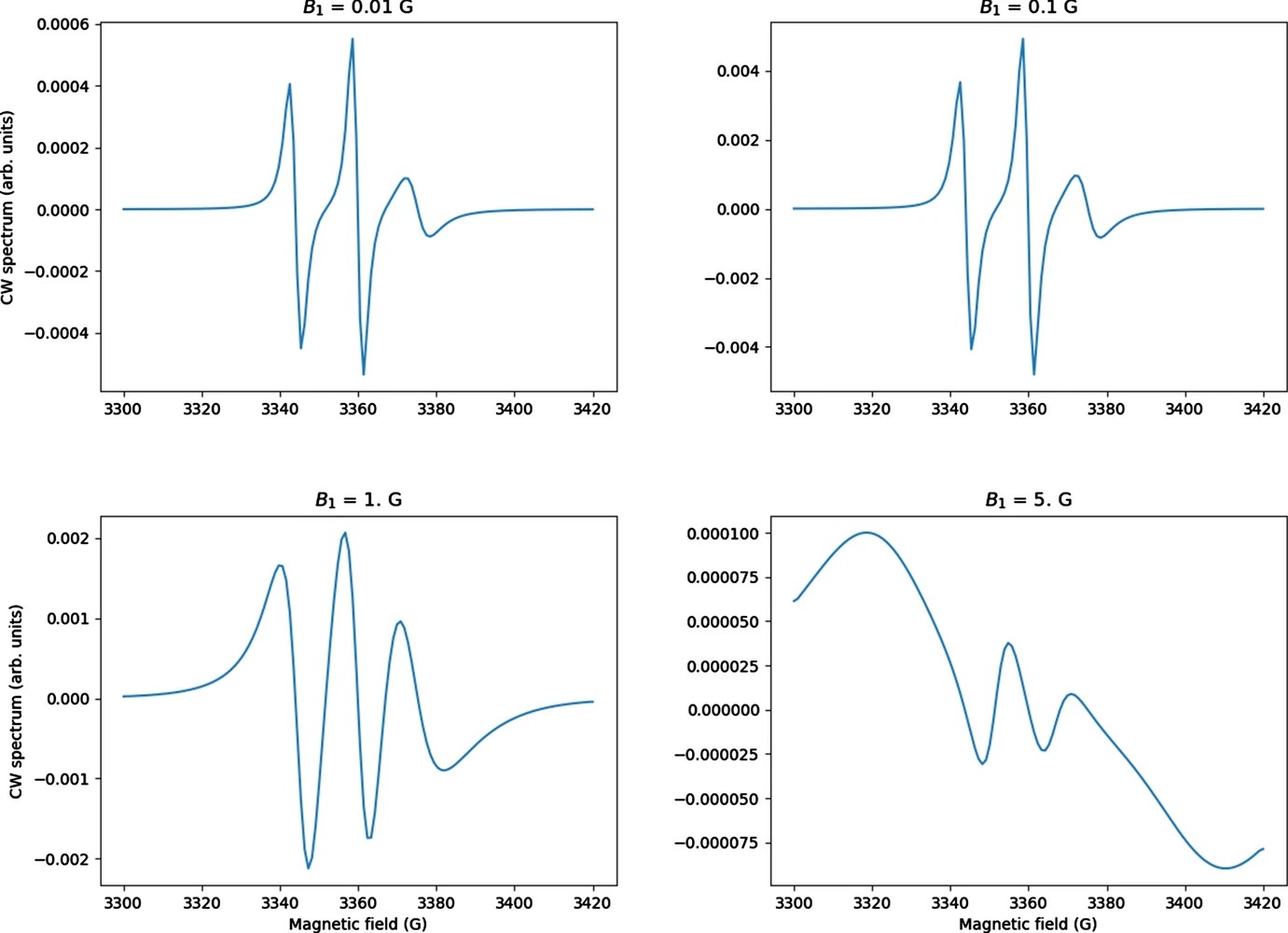
ABSTRACT: CW saturation experiments are widely used in ESR studies of relaxation processes in proteins and lipids. We develop the theory of saturation in ESR spectra in terms of its close relation with that of 2D‐ELDOR. Our treatment of saturation is then based on the microscopic order macroscopic disorder (MOMD) model and can be used to fit the full CW saturation spectrum, rather than fitting just the peak–peak amplitude as a function of microwave field B1 as is commonly done. This requires fewer experiments to yield effects on T1, as well as provides a more extensive dynamic structural picture, for example, for scanning experiments on different protein sites. The code is released as a publicly available software package in Python that can be used to fit CW saturation spectra from biological samples of interest.
|
|
|
Dph3 Enables Aerobic Diphthamide Biosynthesis by Donating One Iron Atom to Transform a [3Fe-4S] to a [4Fe-4S] Cluster in Dph1-Dph2
Y. Zhang, D. Su, B. Dzikovski, S. H. Majer, R. Coleman, S. Chandrasekaran, M. K. Fenwick, B. R. Crane, K. M. Lancaster, J. H. Freed, and H. Lin.
J. Am. Chem. Soc. 143, 9314-9319 (2021)
|

|
|
Dph3 Enables Aerobic Diphthamide Biosynthesis by Donating One Iron Atom to Transform a [3Fe-4S] to a [4Fe-4S] Cluster in Dph1-Dph2
Y. Zhang, D. Su, B. Dzikovski, S. H. Majer, R. Coleman, S. Chandrasekaran, M. K. Fenwick, B. R. Crane, K. M. Lancaster, J. H. Freed, and H. Lin.
J. Am. Chem. Soc. 143, 9314-9319 (2021)
Supporting Information
<doi: 10.1021/jacs.1c03956>
PMID:
34154323
PMCID:
PMC8251694
|
|
|
ABSTRACT: All radical S‐adenosylmethionine (radical‐SAM) enzymes, including the noncanonical radical‐SAM enzyme diphthamide biosynthetic enzyme Dph1–Dph2, require at least one [4Fe–4S](Cys)3 cluster for activity. It is well‐known in the radical‐SAM enzyme community that the [4Fe–4S](Cys)3 cluster is extremely air‐sensitive and requires strict anaerobic conditions to reconstitute activity in vitro. Thus, how such enzymes function in vivo in the presence of oxygen in aerobic organisms is an interesting question. Working on yeast Dph1–Dph2, we found that consistent with the known oxygen sensitivity, the [4Fe–4S] cluster is easily degraded into a [3Fe–4S] cluster. Remarkably, the small iron‐containing protein Dph3 donates one Fe atom to convert the [3Fe–4S] cluster in Dph1–Dph2 to a functional [4Fe–4S] cluster during the radical‐SAM enzyme catalytic cycle. This mechanism to maintain radical‐SAM enzyme activity in aerobic environments is likely general, and Dph3‐like proteins may exist to keep other radical‐SAM enzymes functional in aerobic environments.
|
|
|
Extraction of Weak Spectroscopic Signals with High Fidelity: Examples from ESR
M. Srivastava, B. Dzikovski, and J. H. Freed.
J. Phys. Chem. A 125, 4480-4487 (2021)
Supporting Information
<doi: 10.1021/acs.jpca.1c02241>
PMID:
34009996
PMCID:
PMC8317606
|
|
|
ABSTRACT: Noise impedes experimental studies by reducing signal resolution and/or suppressing weak signals. Signal averaging and filtering are the primary methods used to reduce noise, but they have limited effectiveness and lack capabilities to recover signals at low signal‐to‐noise ratios (SNRs). We utilize a wavelet transform‐based approach to effectively remove noise from spectroscopic data. The wavelet denoising method we use is a significant improvement on standard wavelet denoising approaches. We demonstrate its power in extracting signals from noisy spectra on a variety of signal types ranging from hyperfine lines to overlapped peaks to weak peaks overlaid on strong ones, drawn from electron‐spin‐resonance spectroscopy. The results show that one can accurately extract details of complex spectra, including retrieval of very weak ones. It accurately recovers signals at an SNR of ˜1 and improves the SNR by about 3 orders of magnitude with high fidelity. Our examples show that one is now able to address weaker SNR signals much better than by previous methods. This new wavelet approach can be successfully applied to other spectroscopic signals.
|
|
|
SARS-CoV-2 Fusion Peptide has a Greater Membrane Perturbating Effect than SARS-CoV with Highly Specific Dependence on Ca2+
A. L. Lai and J. H. Freed.
J. Mol. Biol. 433, 166946 (2021)
<doi: 10.1016/j.jmb.2021.166946>
PMID:
33744314
PMCID:
PMC7969826
|

|
|
ABSTRACT: Coronaviruses are a major infectious disease threat, and include the zoonotic‐origin human pathogens SARS‐CoV‐2, SARS‐CoV, and MERS‐CoV (SARS‐2, SARS‐1, and MERS). Entry of coronaviruses into host cells is mediated by the spike (S) protein. In our previous ESR studies, the local membrane ordering effect of the fusion peptide (FP) of various viral glycoproteins including the S of SARS‐1 and MERS has been consistently observed. We previously determined that the sequence immediately downstream from the S2′ cleavage site is the bona fide SARS‐1 FP. In this study, we used sequence alignment to identify the SARS‐2 FP, and studied its membrane ordering effect. Although there are only three residue differences, SARS‐2 FP induces even greater membrane ordering than SARS‐1 FP, possibly due to its greater hydrophobicity. This may be a reason that SARS‐2 is better able to infect host cells. In addition, the membrane binding enthalpy for SARS‐2 is greater. Both the membrane ordering of SARS‐2 and SARS‐1 FPs are dependent on Ca2+, but that of SARS‐2 shows a greater response to the presence of Ca2+. Both FPs bind two Ca2+ ions as does SARS‐1 FP, but the two Ca2+ binding sites of SARS‐2 exhibit greater cooperativity. This Ca2+ dependence by the SARS‐2 FP is very ion‐specific. These results show that Ca2+ is an important regulator that interacts with the SARS‐2 FP and thus plays a significant role in SARS‐2 viral entry. This could lead to therapeutic solutions that either target the FP‐calcium interaction or block the Ca2+ channel.
|
|
|
Microsecond dynamics in proteins by two-dimensional ESR. II. Addressing computational challenges
P. Gupta, K. Chaudhari, and J. H. Freed.
J. Chem. Phys. 154, 084115 (2021)
Supporting Information
<doi: 10.1063/5.0042441>
PMID:
33639766
PMCID:
PMC7928224
|
|
|
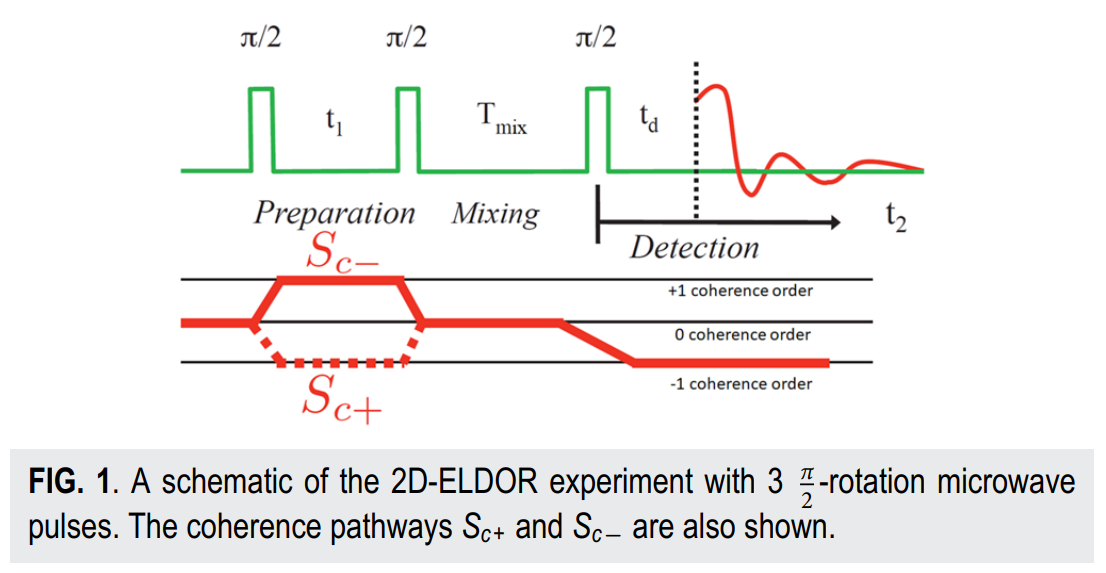
ABSTRACT: Two‐dimensional electron‐electron double resonance (2D‐ELDOR) provides extensive insight into molecular motions. Recent developments permitting experiments at higher frequencies (95 GHz) provide molecular orientational resolution, enabling a clearer description of the nature of the motions. In previous work, we provided simulations for the case of domain motions within proteins that are themselves slowly tumbling in a solution. In order to perform these simulations, it was found that the standard approach of solving the relevant stochastic Liouville equation using the efficient Lanczos algorithm for this case breaks down, so algorithms were employed that rely on the Arnoldi iteration. While they lead to accurate simulations, they are very time‐consuming. In this work, we focus on a variant known as the rational Arnoldi algorithm. We show that this can achieve a significant reduction in computation time. The stochastic Liouville matrix, which is of very large dimension, N, is first reduced to a much smaller dimension, m, e.g., from N ˜ O(104) to m ˜ 60, that spans the relevant Krylov subspace from which the spectrum is predicted. This requires the selection of the m frequency shifts to be utilized. A method of adaptive shift choice is introduced to optimize this selection. We also find that these procedures help in optimizing the pruning procedure that greatly reduces the dimension of the initial N dimensional stochastic Liouville matrix in such subsequent computations.
|
|
|
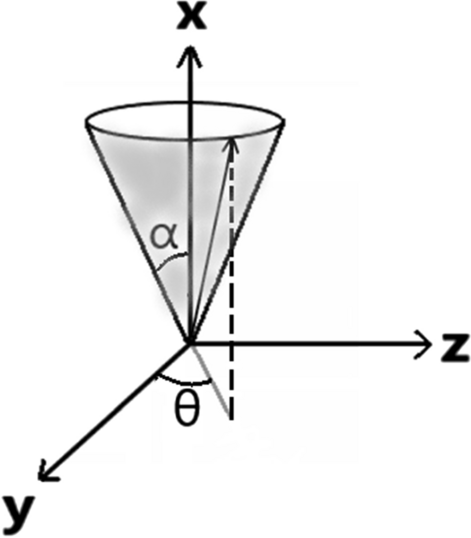
Relaxation in Pulsed EPR: Thermal Fluctuation of Spin-Hamiltonian Parameters of an Electron-Nuclear Spin-Coupled System in a Malonic Acid Single Crystal in a Strong Harmonic-Oscillator Restoring Potential
S. K. Misra and H. R. Salahi
Appl. Magn. Res. 52, 247-261 (2021)
|
|
|
Relaxation in Pulsed EPR: Thermal Fluctuation of Spin-Hamiltonian Parameters of an Electron-Nuclear Spin-Coupled System in a Malonic Acid Single Crystal in a Strong Harmonic-Oscillator Restoring Potential
S. K. Misra and H. R. Salahi
Appl. Magn. Res. 52, 247-261 (2021)
<doi: 10.1007/s00723-020-01308-9>
|
|
|
ABSTRACT: Random fluctuations in the g̃ and à matrices of a spin system due to thermal motion of a molecule are specifically considered to calculate the relaxation matrix for the four-level electron-nuclear spin-coupled system (S = 1/2; I = 1/2) in a malonic acid crystal, using the formalism outlined by Lee et al. (J Chem Phys 98:3665–3689, 1993). The correlation time, τc, and the value of the parameter λ, characterizing the harmonic-oscillator restoring potential of the small-amplitude fluctuation of the director of the malonic-acid molecule due to thermal motion are estimated from the knowledge of the experimental values of (τc, λ). The four electronic, T2e and the two nuclear, Tn, spin relaxation times are calculated to be functions of (τc, λ) governing the fluctuations. The values of (τc, λ), evaluated with these expressions, when fitted to the experimental values of T2e and T2n, assuming the molecule to be in the ground state (n = 0) in the harmonic-oscillator potential, a rather narrow region of (τc, λ) values about τc = 0.081 μs and λ = 4.4 is found. These values are then used to calculate the time-dependent echo-ELDOR signal by the relevant Liouville von-Neumann (LVN) equation. The resulting Fourier transform is found to be in excellent agreement with the experimental data. The (τc, λ) values for the excited states described by n = 1, 2 have also been calculated, although these states are unlikely to be populated at room temperature.
|
|
|
Microsecond Exchange Processes Studied by Two‐Dimensional ESR at 95 GHz
B. Dzikovski, V. V. Khramtsov, S. Chandrasekaran, C. Dunnam, M. Shah, and J. H. Freed.
J. Am. Chem. Soc. 142, 21368-21381 (2020)
Supporting Information
<doi: 10.1021/jacs.0c09469>
PMID:
33305945
PMCID:
PMC7810061
|
|
|
ABSTRACT: Exchange processes which include conformational change, protonation/deprotonation, and binding equilibria are routinely studied by 2D exchange NMR techniques, where information about the exchange of nuclei between environments with different NMR shifts is obtained from the development of cross‐peaks. Whereas 2D NMR enables the real time study of millisecond and slower exchange processes, 2D ESR in the form of 2D‐ELDOR (two‐dimensional electron‐electron double resonance) has the potential for such studies over the nanosecond to microsecond real time scales. Cross‐peak development due to chemical exchange has been seen previously for semiquinones in ESR, but this is not possible for most common ESR probes, such as nitroxides, studied at typical ESR frequencies because, unlike NMR, the exchanging states yield ESR signals that are not resolved from each other within their respective line widths. But at 95 GHz, it becomes possible to resolve them in many cases because of the increased g‐factor resolution. The 95 GHz instrumental developments occurring at ACERT now enable such studies. We demonstrate these new capabilities in two studies: (A) the protonation/deprotonation process for a pH‐sensitive imidazoline spin label in aqueous solution where the exchange rate and the population ratio of the exchanging states are controlled by the concentration and pH of the buffer solution, respectively, and (B) a nitroxide radical partitioning between polar (aqueous) and nonpolar (phospholipid) environments in multilamellar lipid vesicles, where the cross‐peak development arises from the exchange of the nitroxide between the two phases. This work represents the first example of the observation and analysis of cross‐peaks arising from chemical exchange processes involving nitroxide spin labels.
|
|
|
Engineered chemotaxis core signaling units indicate a constrained kinase-off state
A. R. Muok, T. K. Chua, M. Srivastava, W. Yang, Z. Maschmann, P. P. Borbat, J. Chong, S. Zhang, J. H. Freed, A. Briegel, B. R. Crane.
Sci. Signal. 13, eabc1328 (2020)
|
|
|
Engineered chemotaxis core signaling units indicate a constrained kinase-off state
A. R. Muok, T. K. Chua, M. Srivastava, W. Yang, Z. Maschmann, P. P. Borbat, J. Chong, S. Zhang, J. H. Freed, A. Briegel, B. R. Crane.
Sci. Signal. 13, eabc1328 (2020)
Supporting Information
<doi: 10.1126/scisignal.abc1328>
PMID:
33172954
PMCID:
PMC7790435
|
|
|
ABSTRACT: Bacterial chemoreceptors, the histidine kinase CheA, and the coupling protein CheW form transmembrane molecular arrays with remarkable sensing properties. The receptors inhibit or stimulate CheA kinase activity depending on the presence of attractants or repellants, respectively. We engineered chemoreceptor cytoplasmic regions to assume a trimer of receptor dimers configuration that formed well‐defined complexes with CheA and CheW and promoted a CheA kinase‐off state. These mimics of core signaling units were assembled to homogeneity and investigated by site‐directed spin‐labeling with pulse‐dipolar electron‐spin resonance spectroscopy (PDS), small‐angle x‐ray scattering, targeted protein cross‐linking, and cryo–electron microscopy. The kinase‐off state was especially stable, had relatively low domain mobility, and associated the histidine substrate and docking domains with the kinase core, thus preventing catalytic activity. Together, these data provide an experimentally restrained model for the inhibited state of the core signaling unit and suggest that chemoreceptors indirectly sequester the kinase and substrate domains to limit histidine autophosphorylation.
|
|
|
Characterizing Enzyme Reactions in Microcrystals for Effective Mix-and-Inject Experiments using X-ray Free-Electron Lasers
G. D. Calvey, A. M. Katz, K. A. Zielinski, B. Dzikovski, and L. Pollack
Anal. Chem. 92, 13864-13870 (2020)
Supporting Information
<doi: 10.1021/acs.analchem.0c02569>
PMID:
32955854
PMCID:
PMC8367009
|

|
|
ABSTRACT: Mix-and-inject serial crystallography is an emerging technique that utilizes X-ray free-electron lasers (XFELs) and microcrystalline samples to capture atomically detailed snapshots of biomolecules as they function. Early experiments have yielded exciting results; however, there are limited options to characterize reactions in crystallo in advance of the beamtime. Complementary measurements are needed to identify the best conditions and timescales for observing structural intermediates. Here, we describe the interface of XFEL compatible mixing injectors with rapid freeze-quenching and X-band EPR spectroscopy, permitting characterization of reactions in crystals under the same conditions as an XFEL experiment. We demonstrate this technology by tracking the reaction of azide with microcrystalline myoglobin, using only a fraction of the sample required for a mix-and-inject experiment. This spectroscopic method enables optimization of sample and mixer conditions to maximize the populations of intermediate states, eliminating the guesswork of current mix-and-inject experiments.
|
|
|
High-yield production in E. coli and characterization of full-length functional p13II protein from human T-cell leukemia virus type 1
E. R. Georgieva, P. P. Borbat, C. Fanouraki, J. H. Freed.
Protein Expr. Purif. 173, 105659 (2020)
Supporting Information
<doi: 10.1016/j.pep.2020.105659>
PMID:
32360379
PMCID:
PMC7266171
|
|
|
ABSTRACT: Human T-cell leukemia virus type 1 is an oncovirus that causes aggressive adult T-cell leukemia but is also responsible for severe neurodegenerative and endocrine disorders. Combatting HTLV-1 infections requires a detailed understanding of the viral mechanisms in the host. Therefore, in vitro studies of important virus-encoded proteins would be critical. Our focus herein is on the HTLV-1-encoded regulatory protein p13II, which interacts with the inner mitochondrial membrane, increasing its permeability to cations (predominantly potassium, K+). Thereby, this protein affects mitochondrial homeostasis. We report on our progress in developing specific protocols for heterologous expression of p13II in E. coli, and methods for its purification and characterization. We succeeded in producing large quantities of highly-pure full-length p13II, deemed to be its fully functional form. Importantly, our particular approach based on the fusion of ubiquitin to the p13II C-terminus was instrumental in increasing the persistently low expression of soluble p13II in its native form. We subsequently developed approaches for protein spin labeling and a conformation study using double electron-electron resonance (DEER) spectroscopy and a fluorescence-based cation uptake assay for p13II in liposomes. Our DEER results point to large protein conformation changes occurring upon transition from the soluble to the membrane-bound state. The functional assay on p13II-assisted transport of thallium (Tl+) through the membrane, wherein Tl+ substituted for K+, suggests transmembrane potential involvement in p13II function. Our study lays the foundation for expansion of in vitro functional and structural investigations on p13II and would aid in the development of structure-based protein inhibitors and markers.
|
|
|
How cholesterol stiffens unsaturated lipid membranes
S. Chakraborty, M. Doktorova, T. R. Molugu, F. A. Heberle, H. L. Scott, B. Dzikovski, M. Nagao, L.-R. Stingaciu, R. F. Standaert, F. N. Barrera, J. Katsaras, G. Khelashvili, M. F Brown, R. Ashkar
PNAS 117 21896-21905 (2020)
|
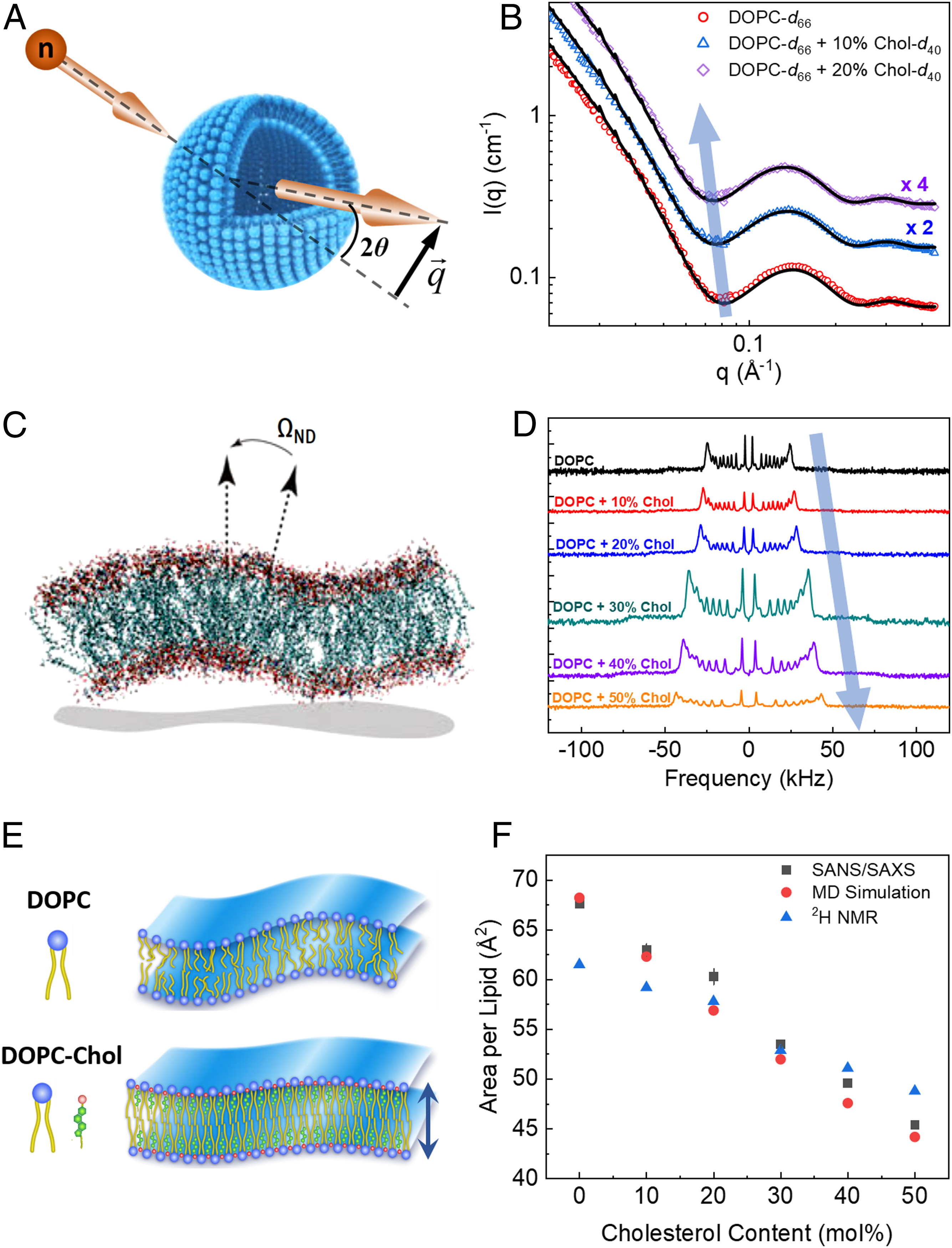
|
|
How cholesterol stiffens unsaturated lipid membranes
S. Chakraborty, M. Doktorova, T. R. Molugu, F. A. Heberle, H. L. Scott, B. Dzikovski, M. Nagao, L.-R. Stingaciu, R. F. Standaert, F. N. Barrera, J. Katsaras, G. Khelashvili, M. F Brown, R. Ashkar
PNAS 117 21896-21905 (2020)
Supporting Information
<doi: 10.1073/pnas.2004807117>
PMID:
32843347
PMCID:
PMC7486787
|
|
|
SIGNIFICANCE: Cholesterol regulates critical cell functions, including lysis, viral budding, and antibiotic resistance, by modifying the bending rigidity of cell membranes; i.e., the ability of membranes to bend or withstand mechanical stresses. A molecular-level understanding of these functions requires knowledge of how cholesterol modifies membrane mechanics over relevant length and time scales. Currently, it is widely accepted that cholesterol has no effect on the mechanical properties of unsaturated lipid membranes, implying that viruses, for example, can bud from regions enriched in (poly)unsaturated lipids. Our observations that cholesterol causes local stiffening in DOPC membranes indicate that a reassessment of existing concepts is necessary. These findings have far-reaching implications in understanding cholesterol's role in biology and its applications in bioengineering and drug design.
ABSTRACT: Cholesterol is an integral component of eukaryotic cell membranes and a key molecule in controlling membrane fluidity, organization, and other physicochemical parameters. It also plays a regulatory function in antibiotic drug resistance and the immune response of cells against viruses, by stabilizing the membrane against structural damage. While it is well understood that, structurally, cholesterol exhibits a densification effect on fluid lipid membranes, its effects on membrane bending rigidity are assumed to be nonuniversal; i.e., cholesterol stiffens saturated lipid membranes, but has no stiffening effect on membranes populated by unsaturated lipids, such as 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC). This observation presents a clear challenge to structure–property relationships and to our understanding of cholesterol-mediated biological functions. Here, using a comprehensive approach–combining neutron spin-echo (NSE) spectroscopy, solid-state deuterium NMR (2H NMR) spectroscopy, and molecular dynamics (MD) simulations–we report that cholesterol locally increases the bending rigidity of DOPC membranes, similar to saturated membranes, by increasing the bilayer's packing density. All three techniques, inherently sensitive to mesoscale bending fluctuations, show up to a threefold increase in effective bending rigidity with increasing cholesterol content approaching a mole fraction of 50%. Our observations are in good agreement with the known effects of cholesterol on the area-compressibility modulus and membrane structure, reaffirming membrane structure–property relationships. The current findings point to a scale-dependent manifestation of membrane properties, highlighting the need to reassess cholesterol's role in controlling membrane bending rigidity over mesoscopic length and time scales of important biological functions, such as viral budding and lipid–protein interactions.
|
|
|
Structural Dynamics by NMR in the Solid State: The Unified MOMD Perspective Applied to Organic Frameworks with Interlocked Molecules
E. Meirovitch, Z. Liang, and J. H. Freed.
J. Phys. Chem. B 124, 6225-6235 (2020)
Supporting Information
<doi: 10.1021/acs.jpcb.0c03687>
PMID:
32584038
PMCID:
PMC7666760
|
|
|
ABSTRACT: The microscopic-order-macroscopic-disorder (MOMD) approach for NMR lineshape analysis has been applied to the University of Windsor Dynamic Materials (UWDM) of types 1, 2, α-3, β-3, and 5, which are metal–organic frameworks (MOFs) comprising mobile mechanically interlocked molecules (MIMs). The mobile MIM components are selectively deuterated crown ether macrocycles – 24C6, 22C6, and B24C6. Their motion is described in MOMD by an effective/collective dynamic mode characterized by a diffusion tensor, R, a restricting/ordering potential, u, expanded in the Wigner rotation matrix elements, D0,KL, and features of local geometry. Experimental 2H lineshapes are available over 220 K (on average) and in some cases 320 K. They are reproduced with axial R, u given by the terms D0,02 and D0,|2|2, and established local geometry. For UWDM of types 1, β-3, and 5, where the macrocycle resides in a relatively loose space, u is in the 1–3 kT, R∥ in the (1.0–2.5) × 106 s–1, and R⊥ in the (0.4–2.5) × 104 s–1 range; the deuterium atom is bonded to a carbon atom with tetrahedral coordination character. For UWDM of types 2 and α-3, where the macrocycle resides in a much tighter space, a substantial change in the symmetry of u and the coordination character of the 2H-bonded carbon are detected at higher temperatures. The activation energies for R∥ and R⊥ are characteristic of each system. The MOMD model is general; effective/collective dynamic modes are treated. The characteristics of motion, ordering, and geometry are physically well-defined; they differ from case to case in extent and symmetry but not in essence. Physical clarity and consistency provide new insights. A previous interpretation of the same experimental data used models consisting of collections of independent simple motions. These models are specific to each case and temperature. Within their scope, generating consistent physical pictures and comparing cases are difficult; possible collective modes are neglected.
|
|
|
Ca2+ Ions Promote Fusion of Middle East Respiratory Syndrome Coronavirus with Host Cells and Increase Infectivity
M. R. Straus, T. Tang, A. L. Lai, A. Flegel, M. Bidon, J. H. Freed, S. Daniel, G. R. Whittaker.
J. Virol. 94, e00426-20 (2020)
Supporting Information
<doi: 10.1128/JVI.00426-20>
PMID:
32295925
PMCID:
PMC7307142
|
|
|
ABSTRACT: Fusion with, and subsequent entry into, the host cell is one of the critical steps in the life cycle of enveloped viruses. For Middle East respiratory syndrome coronavirus (MERS-CoV), the spike (S) protein is the main determinant of viral entry. Proteolytic cleavage of the S protein exposes its fusion peptide (FP), which initiates the process of membrane fusion. Previous studies on the related severe acute respiratory syndrome coronavirus (SARS-CoV) FP have shown that calcium ions (Ca2+) play an important role in fusogenic activity via a Ca2+ binding pocket with conserved glutamic acid (E) and aspartic acid (D) residues. SARS-CoV and MERS-CoV FPs share a high sequence homology, and here, we investigated whether Ca2+ is required for MERS-CoV fusion by screening a mutant array in which E and D residues in the MERS-CoV FP were substituted with neutrally charged alanines (A). Upon verifying mutant cell surface expression and proteolytic cleavage, we tested their ability to mediate pseudoparticle (PP) infection of host cells in modulating Ca2+ environments. Our results demonstrate that intracellular Ca2+ enhances MERS-CoV wild-type (WT) PP infection by approximately 2-fold and that E891 is a crucial residue for Ca2+ interaction. Subsequent electron spin resonance (ESR) experiments revealed that this enhancement could be attributed to Ca2+ increasing MERS-CoV FP fusion-relevant membrane ordering. Intriguingly, isothermal calorimetry showed an approximate 1:1 MERS-CoV FP to Ca2+ ratio, as opposed to an 1:2 SARS-CoV FP to Ca2+ ratio, suggesting significant differences in FP Ca2+ interactions of MERS-CoV and SARS-CoV FP despite their high sequence similarity.
IMPORTANCE Middle East respiratory syndrome coronavirus (MERS-CoV) is a major emerging infectious disease with zoonotic potential and has reservoirs in dromedary camels and bats. Since its first outbreak in 2012, the virus has repeatedly transmitted from camels to humans, with 2,468 confirmed cases causing 851 deaths. To date, there are no efficacious drugs and vaccines against MERS-CoV, increasing its potential to cause a public health emergency. In order to develop novel drugs and vaccines, it is important to understand the molecular mechanisms that enable the virus to infect host cells. Our data have found that calcium is an important regulator of viral fusion by interacting with negatively charged residues in the MERS-CoV FP region. This information can guide therapeutic solutions to block this calcium interaction and also repurpose already approved drugs for this use for a fast response to MERS-CoV outbreaks.
|
|
|
Microsecond dynamics in proteins by two-dimensional ESR: Predictions
P. Gupta, Z. Liang, and J. H. Freed.
J. Chem. Phys. 152, 214112 (2020)
Supporting Information
<doi: 10.1063/5.0008094>
PMID:
32505151
PMCID:
PMC7863697
|

|
|
ABSTRACT: Two-dimensional electron-electron double resonance (2D-ELDOR) provides extensive insight into molecular motions. Recent developments permitting experiments at higher frequencies (95 GHz) provide molecular orientational resolution, enabling a clearer description of the nature of the motions. In this work, simulations are provided for the example of domain motions within proteins that are themselves slowly tumbling in solution. These show the nature of the exchange cross-peaks that are predicted to develop in real time from such domain motions. However, we find that the existing theoretical methods for computing 2D-ELDOR experiments over a wide motional range begin to fail seriously when applied to very slow motions characteristic of proteins in solution. One reason is the failure to obtain accurate eigenvectors and eigenvalues of the complex symmetric stochastic Liouville matrices describing the experiment when computed by the efficient Lanczos algorithm in the range of very slow motion. Another, perhaps more serious, issue is that these matrices are "non-normal," such that for the very slow motional range even rigorous diagonalization algorithms do not yield the correct eigenvalues and eigenvectors. We have employed algorithms that overcome both these issues and lead to valid 2D-ELDOR predictions even for motions approaching the rigid limit. They are utilized to describe the development of cross-peaks in 2D-ELDOR at 95 GHz for a particular case of domain motion.
|
|
|
Conformational Dynamics in Extended RGD-Containing Peptides
W. R. Lindemann, A. J. Mijalis, J. L. Alonso, P. P. Borbat, J. H. Freed, M. A. Arnaout, B. L. Pentelute, and J. H. Ortony.
Biomacromolecules 21, 2786-2794 (2020)
Supporting Information
<doi: 10.1021/acs.biomac.0c00506>
PMID:
32469507
PMCID:
PMC7388056
|
|
|
ABSTRACT: RGD is a prolific example of a tripeptide used in biomaterials for cell adhesion, but the potency of free or surface-bound RGD tripeptide is orders-of-magnitude less than the RGD domain within natural proteins. We designed a set of peptides with varying lengths, composed of fragments of fibronectin protein whose central three residues are RGD, in order to vary their conformational behavior without changing the binding site's chemical environment. With these peptides, we measure the conformational dynamics and transient structure of the active site. Our studies reveal how flanking residues affect conformational behavior and integrin binding. We find that disorder of the binding site is important to the potency of RGD peptides and that transient hydrogen bonding near the RGD site affects both the energy landscape roughness of the peptides and peptide binding. This phenomenon is independent of longer-range folding interactions and helps explain why short binding sequences, including RGD itself, do not fully replicate the integrin-targeting properties of extracellular matrix proteins. Our studies reinforce that peptide binding is a holistic event and fragments larger than those directly involved in binding should be considered in the design of peptide epitopes for functional biomaterials.
|
|
|
George K. Fraenkel, Electron Spin Resonance Pioneer
J.H. Freed.
In Pioneers of Magnetic Resonance. Strom, E. T., Mainz, V. V., Eds. American Chemical Society: Washington, DC, ACS Symposium Series, 2020; Volume 1349, Chapter 8, pp. 137-154.
|
|
|
George K. Fraenkel, Electron Spin Resonance Pioneer
J.H. Freed.
In Pioneers of Magnetic Resonance. Strom, E. T., Mainz, V. V., Eds. American Chemical Society: Washington, DC, ACS Symposium Series, 2020; Volume 1349, Chapter 8, pp. 137-154.
<doi: 10.1021/bk-2020-1349.ch008>
PMID: [None-book] PMCID:
[None-book]
|
|
|
ABSTRACT: George K. Fraenkel (1921-2009), although less known today, was one of the leading pioneers in the development and use of Electron Spin Resonance (ESR) techniques for studying the structure and dynamical interactions of molecules. Together with his students he developed high-sensitivity, high-resolution spectrometers that enabled them to pioneer the study of ESR in free radicals in solution. His work provided breakthroughs that led to advances in several fields of chemistry, and laid the foundations for later research on the properties of biological systems. This chapter provides a thematic overview, arranged chronologically, of his various studies. Short descriptions are provided of his achievements in these areas, based on his most important papers, so that present-day researchers can appreciate the extent of his accomplishments.
|
|
|
Calcium Ions Directly Interact with the Ebola Virus Fusion Peptide To Promote Structure–Function Changes That Enhance Infection
L. Nathan, A. L. Lai, J. K. Millet, M. R. Straus, J. H. Freed, G. R. Whittaker, and S. Daniel.
ACS Infect. Dis. 6, 250-260 (2020)
Supporting Information
<doi: 10.1021/acsinfecdis.9b00296>
PMID:
31746195
PMCID:
PMC7040957
|
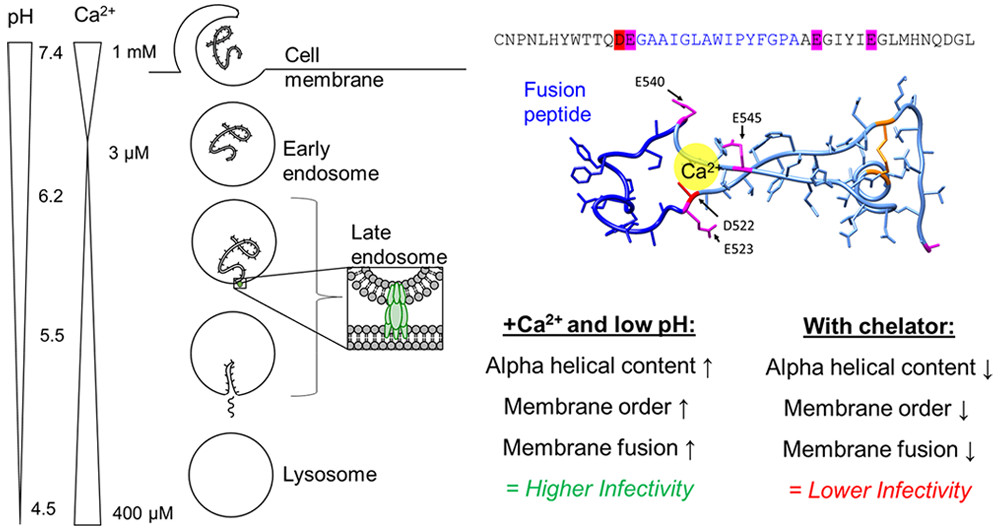
|
|
ABSTRACT: Ebola virus disease is a serious global health concern given its periodic occurrence, high lethality, and the lack of approved therapeutics. Certain drugs that alter intracellular calcium, particularly in endolysosomes, have been shown to inhibit Ebola virus infection; however, the underlying mechanism is unknown. Here, we provide evidence that Zaire ebolavirus (EBOV) infection is promoted in the presence of calcium as a result of the direct interaction of calcium with the EBOV fusion peptide (FP). We identify the glycoprotein residues D522 and E540 in the FP as functionally critical to EBOV's interaction with calcium. We show using spectroscopic and biophysical assays that interactions of the fusion peptide with Ca2+directly targets the Ebola virus fusion peptide and influences its conformation. As these residues are highly conserved across the Filoviridae, calcium's impact on fusion, and subsequently infectivity, is a key interaction that can be leveraged for developing strategies to defend against Ebola infection. This mechanistic insight provides a rationale for the use of calcium-interfering drugs already approved by the FDA as therapeutics against Ebola and enables further development of novel drugs to combat the virus.
|
|
|
Tuning Radical Relay Residues by Proton Management Rescues Protein Electron Hopping
E. F. Yee, B. Dzikovski, and B. R. Crane
J. Am. Chem. Soc. 141, 17571-17587 (2019)
Supporting Information
<doi: 10.1021/jacs.9b05715>
PMID:
31603693
PMCID:
PMC7043243
|
|
|
ABSTRACT: Transient tyrosine and tryptophan radicals play key roles in the electron transfer (ET) reactions of photosystem (PS) II, ribonucleotide reductase (RNR), photolyase, and many other proteins. However, Tyr and Trp are not functionally interchangeable, and the factors controlling their reactivity are often unclear. Cytochrome c peroxidase (CcP) employs a Trp191⋅+ radical to oxidize reduced cytochrome c (Cc). Although a Tyr191 replacement also forms a stable radical, it does not support rapid ET from Cc. Here we probe the redox properties of CcP Y191 by non-natural amino acid substitution, altering the ET driving force and manipulating the protic environment of Y191. Higher potential fluorotyrosine residues increase ET rates marginally, but only addition of a hydrogen bond donor to Tyr191⋅ (via Leu232His or Glu) substantially alters activity by increasing the ET rate by nearly 30-fold. ESR and ESEEM spectroscopies, crystallography, and pH-dependent ET kinetics provide strong evidence for hydrogen bond formation to Y191⋅ by His232/Glu232. Rate measurements and rapid freeze quench ESR spectroscopy further reveal differences in radical propagation and Cc oxidation that support an increased Y191⋅ formal potential of ∼200 mV in the presence of E232. Hence, Y191 inactivity results from a potential drop owing to Y191⋅+ deprotonation. Incorporation of a well-positioned base to accept and donate back a hydrogen bond upshifts the Tyr⋅ potential into a range where it can effectively oxidize Cc. These findings have implications for the YZ/YD radicals of PS II, hole-hopping in RNR and cryptochrome, and engineering proteins for long-range ET reactions.
|
|
|
The asymmetric function of Dph1–Dph2 heterodimer in diphthamide biosynthesis
M. Dong, E. E. Dando, I. Kotliar, X. Su, B. Dzikovski, J. H. Freed, and H. Lin.
J. Biol. Inorg. Chem. 24, 777-782 (2019)
<doi: 10.1007/s00775-019-01702-0>
PMID:
31463593
PMCID:
PMC6893874
|

|
|
ABSTRACT: Diphthamide, the target of diphtheria toxin, is a post-translationally modified histidine residue found in archaeal and eukaryotic translation elongation factor 2 (EF2). In the first step of diphthamide biosynthesis, a [4Fe–4S] cluster-containing radical SAM enzyme, Dph1–Dph2 heterodimer in eukaryotes or Dph2 homodimer in archaea, cleaves S-adenosylmethionine and transfers the 3-amino-3-carboxypropyl group to EF2. It was demonstrated previously that for the archaeal Dph2 homodimer, only one [4Fe–4S] cluster is necessary for the in vitro activity. Here, we demonstrate that for the eukaryotic Dph1–Dph2 heterodimer, the [4Fe–4S] cluster-binding cysteine residues in each subunit are required for diphthamide biosynthesis to occur in vivo. Furthermore, our in vitro reconstitution experiments with Dph1–Dph2 mutants suggested that the Dph1 cluster serves a catalytic role, while the Dph2 cluster facilitates the reduction of the Dph1 cluster by the physiological reducing system Dph3/Cbr1/NADH. Our results reveal the asymmetric functional roles of the Dph1–Dph2 heterodimer and may help to understand how the Fe–S clusters in radical SAM enzymes are reduced in biology.
|
|
|
Local ordering and dynamics in anisotropic media by magnetic resonance: from liquid crystals to proteins
E. Meirovitch & J. H. Freed.
Liq. Cryst. 47, 1926-1954 (2020)
<doi: 10.1080/02678292.2019.1622158>
PMID:
32435078
PMCID:
PMC7239324
|
|
|
ABSTRACT: Magnetic resonance methods have been used extensively for over 50 years to elucidate molecular structure and dynamics of liquid crystals (LCs), providing information quite unique in its rigour and extent. The ESR- or NMR-active probe is often a solute molecule reporting on characteristics associated with the surrounding (LC) medium, which exerts the spatial restrictions on the probe. The theoretical approaches developed for LCs are applicable to anisotropic media in general. Of particular interest is the interior space of a globular protein labelled, e.g. with a nitroxide moiety or a 15N–1H bond. The ESR or NMR label plays the role of the probe and the internal protein surroundings the role of the anisotropic medium. A general feature of the restricted motions is the local ordering, i.e. the nature, magnitude and symmetry of the spatial restraints exerted at the site of the moving probe. This property is the main theme of the present review article. We outline its treatment in our work from both the theoretical and the experimental points of view, highlighting the new physical insights gained. Our illustrations include studies on thermotropic (nematic and smectic) and lyotropic liquid crystals formed by phospholipids, in addition to studies of proteins.
|
|
|
CcdB at pH 4 Forms a Partially Unfolded State with a Dry Core
C. Baliga, B. Selmke, I. Worobiew, P. P. Borbat, S. P. Sarma, W. E Trommer, R. Varadarajan, and N. Aghera
Biophys. J. 116, 807-817 (2019)
Supporting Information
<doi: 10.1016/j.bpj.2019.01.026>
PMID:
30777307
PMCID:
PMC6401195
|
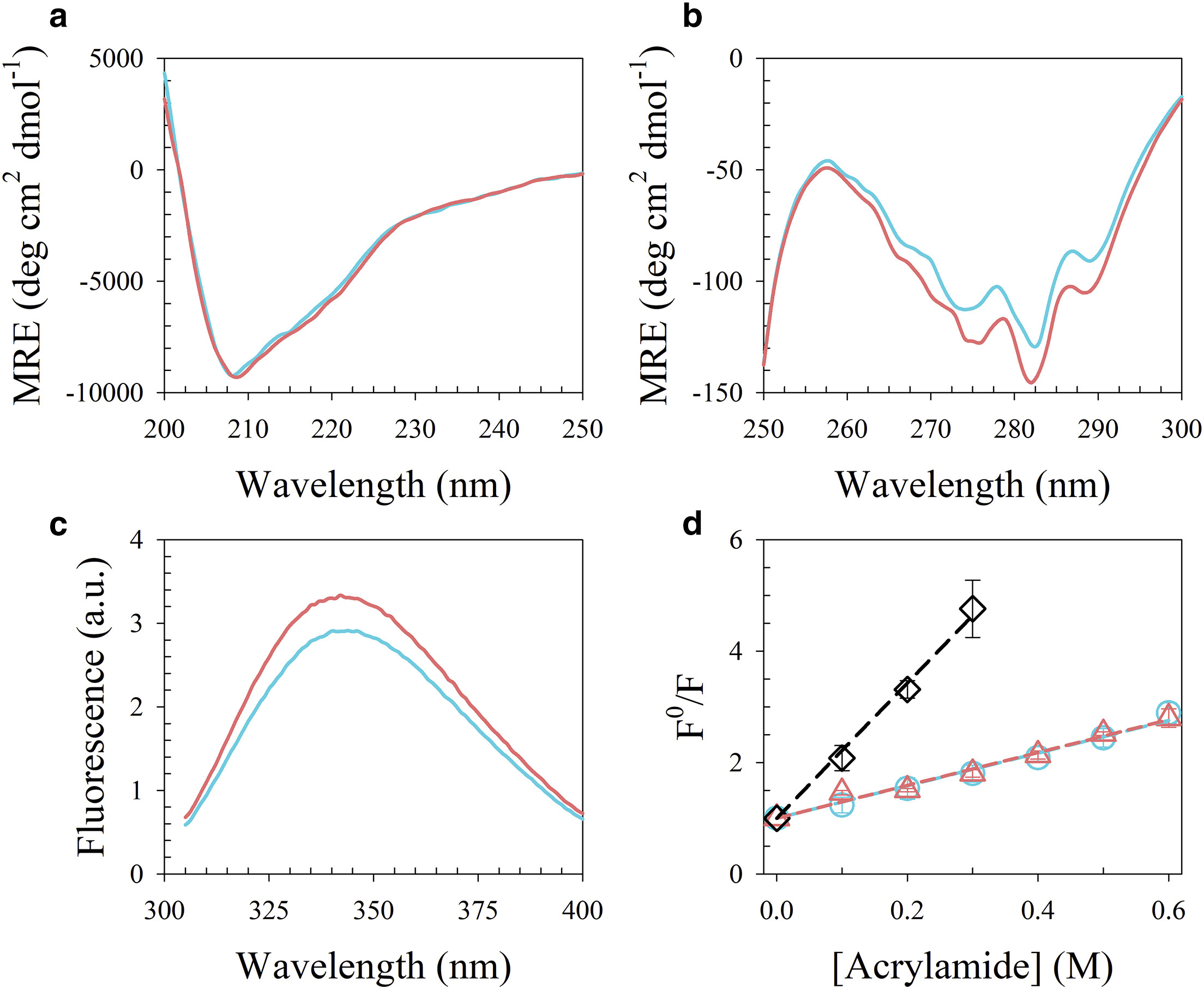
|
|
ABSTRACT: pH is an important factor that affects the protein structure, stability, and activity. Here, we probe the nature of the low-pH structural form of the homodimeric CcdB (controller of cell death B) protein. Characterization of CcdB protein at pH 4 and 300 K using circular dichroism spectroscopy, 8-anilino-1-naphthalene-sulphonate binding, and Trp solvation studies suggests that it forms a partially unfolded state with a dry core at equilibrium under these conditions. CcdB remains dimeric at pH 4 as shown by multiple techniques, such as size-exclusion chromatography coupled to multiangle light scattering, analytical ultracentrifugation, and electron paramagnetic resonance. Comparative analysis using two-dimensional 15N-1H heteronuclear single-quantum coherence NMR spectra of CcdB at pH 4 and 7 suggests that the pH 4 and native state have similar but nonidentical structures. Hydrogen-exchange-mass-spectrometry studies demonstrate that the pH 4 state has substantial but anisotropic changes in local stability with core regions close to the dimer interface showing lower protection but some other regions showing higher protection relative to pH 7.
|
|
|
Insights into histidine kinase activation mechanisms from the monomeric blue light sensor EL346
I. Dikiy, U. R. Edupuganti, R. R. Abzalimov, P. P. Borbat, M. Srivastava, J. H. Freed, and K. H. Gardner.
Proc. Natl. Acad. Sci. 116, 4963-4972 (2019)
Supporting Information
<doi: 10.1073/pnas.1813586116>
PMID:
30808807
PMCID:
PMC6421462
|

|
|
SIGNIFICANCE: All living things must sense and react to their environment. Many single-celled organisms do so by using two-component systems, most simply consisting of a sensor histidine kinase and a response regulator. These systems are involved in pathogenicity pathways and can be targeted by new antibiotics. However, the molecular mechanisms used by histidine kinases to translate sensing into responses are not well understood. To probe this general question, we apply a combination of biophysical techniques to a monomeric histidine kinase that senses blue light to determine the structural changes occurring upon activation. We find these changes to be similar to those predicted for the common dimeric histidine kinases, illustrating that the mechanism of activation is conserved regardless of oligomeric state.
ABSTRACT: Translation of environmental cues into cellular behavior is a necessary process in all forms of life. In bacteria, this process frequently involves two-component systems in which a sensor histidine kinase (HK) autophosphorylates in response to a stimulus before subsequently transferring the phosphoryl group to a response regulator that controls downstream effectors. Many details of the molecular mechanisms of HK activation are still unclear due to complications associated with the multiple signaling states of these large, multidomain proteins. To address these challenges, we combined complementary solution biophysical approaches to examine the conformational changes upon activation of a minimal, blue-light–sensing histidine kinase from Erythrobacter litoralis HTCC2594, EL346. Our data show that multiple conformations coexist in the dark state of EL346 in solution, which may explain the enzyme's residual dark-state activity. We also observe that activation involves destabilization of the helices in the dimerization and histidine phosphotransfer-like domain, where the phosphoacceptor histidine resides, and their interactions with the catalytic domain. Similar light-induced changes occur to some extent even in constitutively active or inactive mutants, showing that light sensing can be decoupled from activation of kinase activity. These structural changes mirror those inferred by comparing X-ray crystal structures of inactive and active HK fragments, suggesting that they are at the core of conformational changes leading to HK activation. More broadly, our findings uncover surprising complexity in this simple system and allow us to outline a mechanism of the multiple steps of HK activation.
|
|
|
|
|
Cofactors are essential constituents of stable and seeding-active tau fibrils
Y. Fichou, Y. Lin, J. N. Rauch, M. Vigers, Z. Zeng, M. Srivastava, T. J. Keller, J. H. Freed, K. S. Kosik, and S. Han.
Proc. Natl. Acad. Sci. 115, 13234-13239 (2018)
|

|
|
Cofactors are essential constituents of stable and seeding-active tau fibrils
Y. Fichou, Y. Lin, J. N. Rauch, M. Vigers, Z. Zeng, M. Srivastava, T. J. Keller, J. H. Freed, K. S. Kosik, and S. Han.
Proc. Natl. Acad. Sci. 115, 13234-13239 (2018)
Supporting Information
<doi: 10.1073/pnas.1810058115>
PMID:
30538196
PMCID:
PMC6310788
|
|
|
SIGNIFICANCE: The tau protein is involved in Alzheimer's and other neurodegenerative diseases, where the location, morphology, and quantity of amyloid fibrils composed of tau correlate with the disease type and stage. While tau fibrillary aggregates have been colocalized in brains together with several cofactors, their role in fibril formation, structure, and seeding has been largely neglected. We show that seeding of tau aggregation is facilitated by polyanionic cofactors, and that seeded or recombinant mature fibrils depolymerize into monomers when their cofactor is removed. We show that cofactor-assisted seeding with mouse brain-derived tau fibrils yielded tau fibrils with distinct and narrowed structural properties compared with heparin-induced fibrils, suggesting that the fibrillar templates tuned the structure of the seeded fibril.
ABSTRACT: Amyloid fibrils are cross-β–rich aggregates that are exceptionally stable forms of protein assembly. Accumulation of tau amyloid fibrils is involved in many neurodegenerative diseases, including Alzheimer's disease (AD). Heparin-induced aggregates have been widely used and assumed to be a good tau amyloid fibril model for most biophysical studies. Here we show that mature fibrils made of 4R tau variants, prepared with heparin or RNA, spontaneously depolymerize and release monomers when their cofactors are removed. We demonstrate that the cross-β-sheet assembly formed in vitro with polyanion addition is unstable at room temperature. We furthermore demonstrate high seeding capacity with transgenic AD mouse brain-extracted tau fibrils in vitro that, however, is exhausted after one generation, while supplementation with RNA cofactors resulted in sustained seeding over multiple generations. We suggest that tau fibrils formed in brains are supported by unknown cofactors and inhere higher-quality packing, as reflected in a more distinct conformational arrangement in the mouse fibril-seeded, compared with heparin-induced, tau fibrils. Our study suggests that the role of cofactors in tauopathies is a worthy focus of future studies, as they may be viable targets for diagnosis and therapeutics.
|
|
|
Singular Value Decomposition Method To Determine Distance Distributions in Pulsed Dipolar Electron Spin Resonance: II. Estimating Uncertainty
M. Srivastava and J. H. Freed.
J. Phys. Chem. A 123, 359-370 (2019)
Supporting Information
<doi: 10.1021/acs.jpca.8b07673>
PMID:
30525624
PMCID:
PMC6372347
|
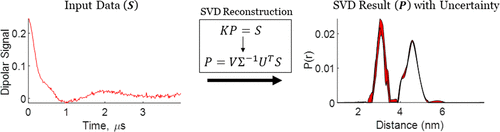
|
|
ABSTRACT: This paper is a continuation of the method introduced by Srivastava and Freed (2017) that is a new method based on truncated singular value decomposition (TSVD) for obtaining physical results from experimental signals without any need for Tikhonov regularization or other similar methods that require a regularization parameter. We show here how to estimate the uncertainty in the SVD-generated solutions. The uncertainty in the solution may be obtained by finding the minimum and maximum values over which the solution remains converged. These are obtained from the optimum range of singular value contributions, where the width of this region depends on the solution point location (e.g., distance) and the signal-to-noise ratio (SNR) of the signal. The uncertainty levels typically found are very small with substantial SNR of the (denoised) signal, emphasizing the reliability of the method. With poorer SNR, the method is still satisfactory but with greater uncertainty, as expected. Pulsed dipolar electron spin resonance spectroscopy experiments are used as an example, but this TSVD approach is general and thus applicable to any similar experimental method wherein singular matrix inversion is needed to obtain the physically relevant result. We show that the Srivastava–Freed TSVD method along with the estimate of uncertainty can be effectively applied to pulsed dipolar electron spin resonance signals with SNR > 30, and even for a weak signal (e.g., SNR ≈ 3) reliable results are obtained by this method, provided the signal is first denoised using wavelet transforms (WavPDS).
|
|
|
Nucleotide Spin Labeling for ESR Spectroscopy of ATP–Binding Proteins
A. R. Muok, T. K. Chua, H. Le and B. R. Crane
Appl. Magn. Res. 49, 1385-1395, (2018)
|

|
|
Nucleotide Spin Labeling for ESR Spectroscopy of ATP–Binding Proteins
A. R. Muok, T. K. Chua, H. Le and B. R. Crane
Appl. Magn. Res. 49, 1385-1395, (2018)
<doi: 10.1007/s00723-018-1070-6>
PMID:
30686862
PMCID:
PMC6342010
|
|
|
ABSTRACT: Site-directed spin labeling of proteins by chemical modification of engineered cysteine residues with the molecule MTSSL (1-oxyl-2,2,5,5-tetramethylpyrroline-3-methyl methanethiosulfonate) has been an invaluable tool for conducting double electron electron resonance (DEER) spectroscopy experiments. However, this method is generally limited to recombinant proteins with a limited number of reactive Cys residues that when modified will not impair protein function. Here, we present a method that allows for spin labeling of protein-nucleotide-binding sites by adenosine diphosphate (ADP) modified with a nitroxide moiety on the β-phosphate (ADP-β-S-SL). The synthesis of this ADP analog is straightforward and isolation of pure product is readily achieved on a standard reverse-phase high-performance liquid chromatography (HPLC) system. Furthermore, analyses of isolated ADP-β-S-SL by LC–mass spectrometry confirm that the molecule is very stable under ambient conditions. The crystal structure of ADP-β-S-SL bound to the ATP pocket of the histidine kinase CheA reveals specific targeting of the probe, whose nitroxide moiety is mobile on the protein surface. Continuous wave and pulsed-ESR measurements demonstrate the capability of ADP-β-S-SL to report on active site environment and provide reliable DEER distance constraints.
|
|
|
Non-Structural Proteins from Human T-cell Leukemia Virus Type 1 in Cellular Membranes—Mechanisms for Viral Survivability and Proliferation
E. R. Georgieva
Int. J. Mol. Sci. 19, 3508 (2018)
<doi: 10.3390/ijms19113508>
PMID:
30413005
PMCID:
PMC6274929
|
|
|
ABSTRACT: Human T-cell leukemia virus type 1 (HTLV-1) is the causative agent of illnesses, such as adult T-cell leukemia/lymphoma, myelopathy/tropical spastic paraparesis (a neurodegenerative disorder), and other diseases. Therefore, HTLV-1 infection is a serious public health concern. Currently, diseases caused by HTLV-1 cannot be prevented or cured. Hence, there is a pressing need to comprehensively understand the mechanisms of HTLV-1 infection and intervention in host cell physiology. HTLV-1-encoded non-structural proteins that reside and function in the cellular membranes are of particular interest, because they alter cellular components, signaling pathways, and transcriptional mechanisms. Summarized herein is the current knowledge about the functions of the membrane-associated p8I, p12I, and p13II regulatory non-structural proteins. p12I resides in endomembranes and interacts with host proteins on the pathways of signal transduction, thus preventing immune responses to the virus. p8I is a proteolytic product of p12I residing in the plasma membrane, where it contributes to T-cell deactivation and participates in cellular conduits, enhancing virus transmission. p13II associates with the inner mitochondrial membrane, where it is proposed to function as a potassium channel. Potassium influx through p13II in the matrix causes membrane depolarization and triggers processes that lead to either T-cell activation or cell death through apoptosis.
|
|
|
Site-Specific Incorporation of a Cu2+ Spin Label into Proteins for Measuring Distances by Pulsed Dipolar Electron Spin Resonance Spectroscopy
G. E. Merz, P. P. Borbat, A. R. Muok, M. Srivastava, D. N. Bunck, J. H. Freed, and B. R. Crane.
J. Phys. Chem. B 122, 9443-9451 (2018)
Supporting Information
<doi: 10.1021/acs.jpcb.8b05619>
PMID:
30222354
PMCID:
PMC6215709
|
|
|
ABSTRACT: Pulsed dipolar electron spin resonance spectroscopy (PDS) is a powerful tool for measuring distances in solution-state macromolecules. Paramagnetic metal ions, such as Cu2+, are used as spin probes because they can report on metalloprotein features and can be spectroscopically distinguished from traditional nitroxide (NO)-based labels. Here, we demonstrate site-specific incorporation of Cu2+ into non-metalloproteins through the use of a genetically encodable non-natural amino acid, 3-pyrazolyltyrosine (PyTyr). We first incorporate PyTyr in cyan fluorescent protein to measure Cu2+-to-NO distances and examine the effects of solvent conditions on Cu2+ binding and protein aggregation. We then apply the method to characterize the complex formed by the histidine kinase CheA and its target response regulator CheY. The X-ray structure of CheY–PyTyr confirms Cu labeling at PyTyr but also reveals a secondary Cu site. Cu2+-to-NO and Cu2+-to-Cu2+ PDS measurements of CheY–PyTyr with nitroxide-labeled CheA provide new insights into the conformational landscape of the phosphotransfer complex and have implications for kinase regulation.
|
|
|
Electrochemical Azidooxygenation of Alkenes Mediated by a TEMPO–N3 Charge-Transfer Complex
J. C. Siu, G. S. Sauer, A. Saha, R. L. Macey, N. Fu, T. Chauviré, K. M. Lancaster, S. Lin
J. Am. Chem. Soc. 140, 12511-12520 (2018)
Supporting Information
<doi: 10.1021/jacs.8b06744>
PMID:
30160949
PMCID:
PMC6212300
|
|
|
ABSTRACT: We report a mild and efficient electrochemical protocol to access a variety of vicinally C–O and C–N difunctionalized compounds from simple alkenes. Detailed mechanistic studies revealed a distinct reaction pathway from those previously reported for TEMPO-mediated reactions. In this mechanism, electrochemically generated oxoammonium ion facilitates the formation of azidyl radical via a charge-transfer complex with azide, TEMPO–N3. DFT calculations together with spectroscopic characterization provided a tentative structural assignment of this charge-transfer complex. Kinetic and kinetic isotopic effect studies revealed that reversible dissociation of TEMPO–N3 into TEMPO⋅ and azidyl precedes the addition of these radicals across the alkene in the rate-determining step. The resulting azidooxygenated product could then be easily manipulated for further synthetic elaborations. The discovery of this new reaction pathway mediated by the TEMPO+/TEMPO⋅ redox couple may expand the scope of aminoxyl radical chemistry in synthetic contexts.
|
|
|
Phenyl-Ring Dynamics in Amyloid Fibrils and Proteins: The Microscopic-Order-Macroscopic-Disorder Perspective
E. Meirovitch, Z. Liang, and J. H. Freed.
J. Phys. Chem. B 122, 8675-8684 (2018)
Supporting Information
<doi: 10.1021/acs.jpcb.8b06330>
PMID:
30141954
PMCID:
PMC6174686
|
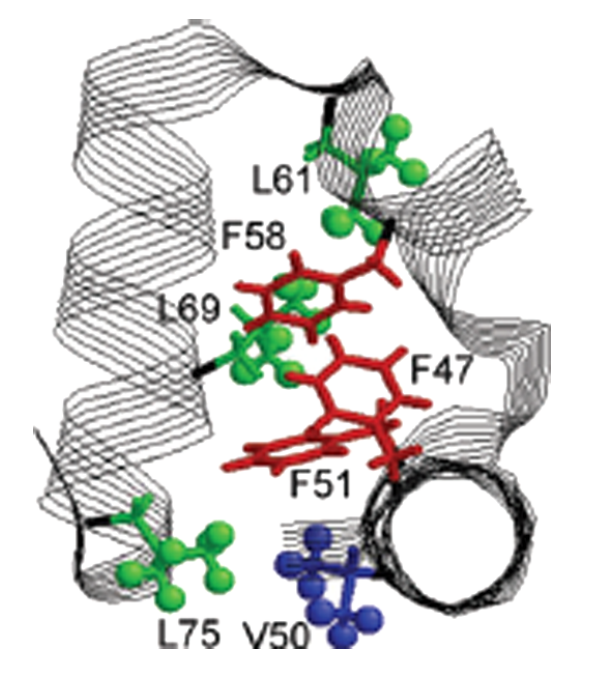
|
|
ABSTRACT: We have developed the microscopic-order-macroscopic-disorder (MOMD) approach for studying internal mobility in polycrystalline proteins with 2H lineshape analysis. The motion itself is expressed by a diffusion tensor, R, the local spatial restraints by a potential, u, and the "local geometry" by the relative orientation of the model-related and nuclear magnetic resonance-related tensors. Here, we apply MOMD to phenyl-ring dynamics in several Αβ40-amyloid-fibrils, and the villin headpiece subdomain (HP36). Because the available data are limited in extent and sensitivity, we adjust u and R in the relevant parameter ranges, fixing the "local geometry" in accordance with standard stereochemistry. This yields a physically well-defined and consistent picture of phenyl-ring dynamics, enabling comparison between different systems. In the temperature range of 278–308 K, u has a strength of (1.7–1.8) kT and a rhombicity of (2.4–2.6) kT, and R has components of 5.0 × 102 ≤ R⊥ ≤ 2.0 × 103 s–1 and 6.3 × 105 ≤ R∥ ≤ 2.0 × 106 s–1. At 278 K, fibril hydration increases the axiality of both u and R; HP36 hydration has a similar effect at 295 K, reducing R⊥ considerably. The D23N mutation slows down the motion of the probe; Αβ40 polymorphism affects both this motion and the related local potential. The present study identifies the impact of various factors on phenyl-ring mobility in amyloid fibrils and globular proteins; the difference between the two protein forms is considerable. The distinctive impact of hydration on phenyl-ring motion and previously studied methyl-group motion is also examined. The 2H lineshapes considered here were analyzed previously with various multi-simple-mode (MSM) models, where several simple motional modes are combined. The MOMD and MSM interpretations differ in essence.
|
|
|
Open and Closed Form of Maltose Binding Protein in Its Native and Molten Globule State As Studied by Electron Paramagnetic Resonance Spectroscopy
B. Selmke, P. P. Borbat, C. Nickolaus, R. Varadarajan, J. H. Freed, and W. E. Trommer.
Biochemistry 57, 5507-5512 (2018)
Supporting Information
<doi: 10.1021/acs.biochem.8b00322>
PMID:
30004675
PMCID:
PMC6211580
|
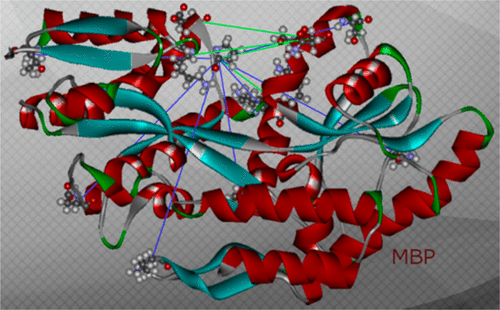
|
|
ABSTRACT: An intensively investigated intermediate state of protein folding is the molten globule (MG) state, which contains secondary but hardly any tertiary structure. In previous work, we have determined the distances between interacting spins within maltose binding protein (MBP) in its native state using continuous wave and double electron–electron resonance (DEER) electron paramagnetic resonance (EPR) spectroscopy. Seven double mutants had been employed to investigate the structure within the two domains of MBP. DEER data nicely corroborated the previously available X-ray data. Even in its MG state, MBP is known to still bind its ligand maltose. We therefore hypothesized that there must be a defined structure around the binding pocket of MBP already in the absence of tertiary structure. Here we have investigated the functional and structural difference between native and MG state in the open and closed form with a new set of MBP mutants. In these, the spin-label positions were placed near the active site. Binding of its ligands leads to a conformational change from open to closed state, where the two domains are more closely together. The complete set of MBP mutants was analyzed at pH 3.2 (MG) and pH 7.4 (native state) using double-quantum coherence EPR. The values were compared with theoretical predictions of distances between the labels in biradicals constructed by molecular modeling from the crystal structures of MBP in open and closed form and were found to be in excellent agreement. Measurements show a defined structure around the binding pocket of MBP in MG, which explains maltose binding. A new and important finding is that in both states ligand-free MBP can be found in open and closed form, while ligand-bound MBP appears only in closed form because of maltose binding.
|
|
|
A facile approach for the in vitro assembly of multimeric membrane transport proteins
E. A. Riederer, P. J. Focke, E. R. Georgieva, N. Akyuz, K. Matulef, P. P. Borbat, J. H. Freed, S. C. Blanchard, O. Boudker, and F. I. Valiyaveetil.
eLife 7, e36478 (2018)
|
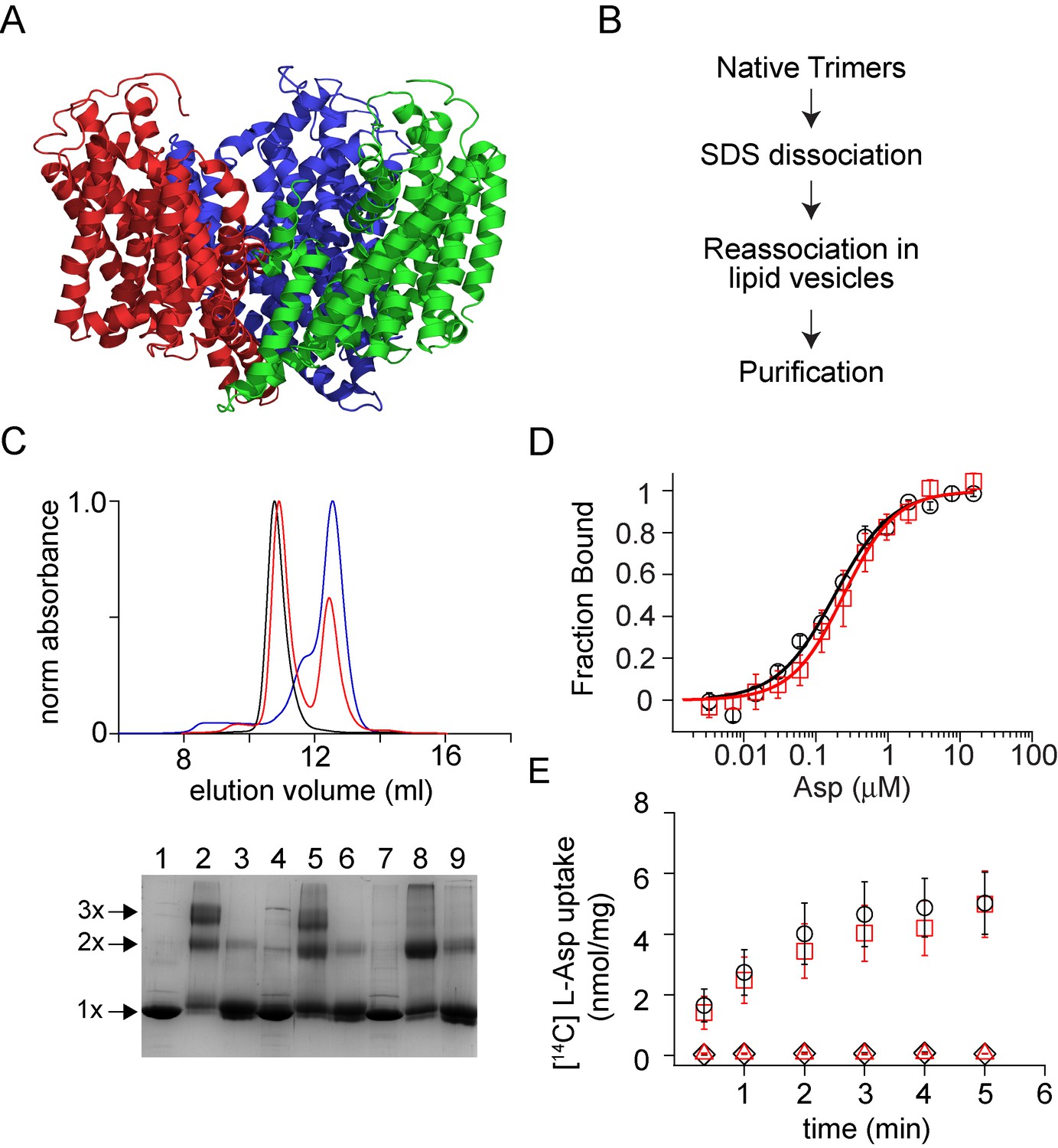
|
|
A facile approach for the in vitro assembly of multimeric membrane transport proteins
E. A. Riederer, P. J. Focke, E. R. Georgieva, N. Akyuz, K. Matulef, P. P. Borbat, J. H. Freed, S. C. Blanchard, O. Boudker, and F. I. Valiyaveetil.
eLife 7, e36478 (2018)
Supporting Information
<doi: 10.7554/eLife.36478>
PMID:
29889023
PMCID:
PMC6025958
|
|
|
ABSTRACT: Membrane proteins such as ion channels and transporters are frequently homomeric. The homomeric nature raises important questions regarding coupling between subunits and complicates the application of techniques such as FRET or DEER spectroscopy. These challenges can be overcome if the subunits of a homomeric protein can be independently modified for functional or spectroscopic studies. Here, we describe a general approach for in vitro assembly that can be used for the generation of heteromeric variants of homomeric membrane proteins. We establish the approach using GltPh, a glutamate transporter homolog that is trimeric in the native state. We use heteromeric GltPh transporters to directly demonstrate the lack of coupling in substrate binding and demonstrate how heteromeric transporters considerably simplify the application of DEER spectroscopy. Further, we demonstrate the general applicability of this approach by carrying out the in vitro assembly of VcINDY, a Na+-coupled succinate transporter and CLC-ec1, a Cl-/H+ antiporter.
|
|
|
Structural basis for membrane anchoring and fusion regulation of the herpes simplex virus fusogen gB
R. S. Cooper, E. R. Georgieva, P. P. Borbat, J. H. Freed, and E. E. Heldwein.
Nat. Struct. Mol. Biol. 25, 416-424 (2018)
Supporting Information
<doi: 10.1038/s41594-018-0060-6>
PMID:
29728654
PMCID:
PMC5942590
|
|
|
ABSTRACT: Viral fusogens merge viral and cell membranes during cell penetration. Their ectodomains drive fusion by undergoing large-scale refolding, but little is known about the functionally important regions located within or near the membrane. Here we report the crystal structure of full-length glycoprotein B (gB), the fusogen from herpes simplex virus, complemented by electron spin resonance measurements. The membrane-proximal (MPR), transmembrane (TMD), and cytoplasmic (CTD) domains form a uniquely folded trimeric pedestal beneath the ectodomain, which balances dynamic flexibility with extensive, stabilizing membrane interactions. The postfusion conformation of the ectodomain suggests that the CTD likewise adopted the postfusion form. However, hyperfusogenic mutations, which destabilize the prefusion state of gB, target key interfaces and structural motifs that reinforce the observed CTD structure. Thus, a similar CTD structure must stabilize gB in its prefusion state. Our data suggest a model for how this dynamic, membrane-dependent 'clamp' controls the fusogenic refolding of gB.
|
|
|
MOMD Analysis of NMR Line Shapes from Aβ-Amyloid Fibrils: A New Tool for Characterizing Molecular Environments in Protein Aggregates
E. Meirovitch, Z. Liang, and J. H. Freed.
J. Phys. Chem. B 122, 4793-4801 (2018)
Supporting Information
<doi: 10.1021/acs.jpcb.8b02181>
PMID:
29624402
PMCID:
PMC5945340
|
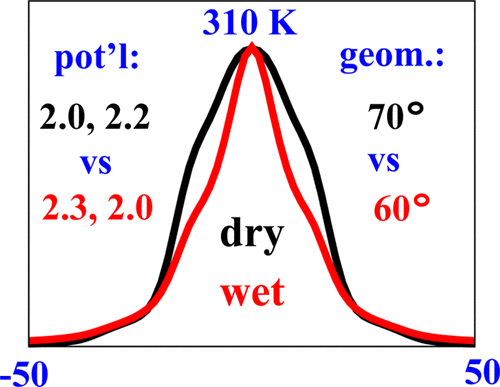
|
|
ABSTRACT: The microscopic-order-macroscopic-disorder (MOMD) approach for 2H NMR line shape analysis is applied to dry and hydrated 3-fold- and 2-fold-symmetric amyloid-Aβ40 fibrils and protofibrils of the D23N mutant. The methyl moieties of L17, L34, V36 (C–CD3), and M35 (S–CD3) serve as probes. Experimental 2H spectra acquired previously in the 147–310 K range are used. MOMD describes local probe motion as axial diffusion (R tensor) in the presence of a potential, u, which represents the spatial restrictions exerted by the molecular surroundings. We find that R∥ = (0.2–3.3) × 104 s–1, R⊥ = (2.2–2.5) × 102 s–1, and R is tilted from the 2H quadrupolar tensor at 60–75°. The strength of u is in the (2.0–2.4) kT range; its rhombicity is substantial. The only methyl moieties affected by fibril hydration are those of M35, located at fibril interfaces. The associated local potentials change form abruptly around 260 K, where massive water freezing occurs. An independent study revealed unfrozen "tightly-peptide-bound" water residing at the interfaces of the 3-fold-symmetric Aβ40 fibrils and at the interfaces of the E22G and E22Δ Aβ40-mutant fibrils. Considering this to be the case in general for Aβ40-related fibrils, the following emerges. The impact of water freezing is transmitted selectively to the fibril structure through interactions with tightly-peptide-bound water, in this case of M35 methyl moieties. The proof that such waters reside at the interfaces of the 2-fold-symmetric fibril, and the protofibril of the D23N mutant, is new. MOMD provides information on the surroundings of the NMR probe directly via the potential, u, which is inherent to the model; a prior interpretation of the same experimental data does so partially and indirectly (see below). Thus, MOMD analysis of NMR line shapes as applied to amyloid fibrils/protein aggregates emerges as a consistent new tool for elucidating the properties of, and processes associated with, molecular environments in the fibril.
|
|
|
Organometallic and radical intermediates reveal mechanism of diphthamide biosynthesis
M. Dong, V. Kathiresan, M. K. Fenwick, A. T. Torelli, Y. Zhang, J. D. Caranto, B. Dzikovski, A. Sharma, K. M. Lancaster, J. H. Freed, S. E. Ealick, B. M. Hoffman, and H. Lin.
Science 359, 1247-1250 (2018)
|
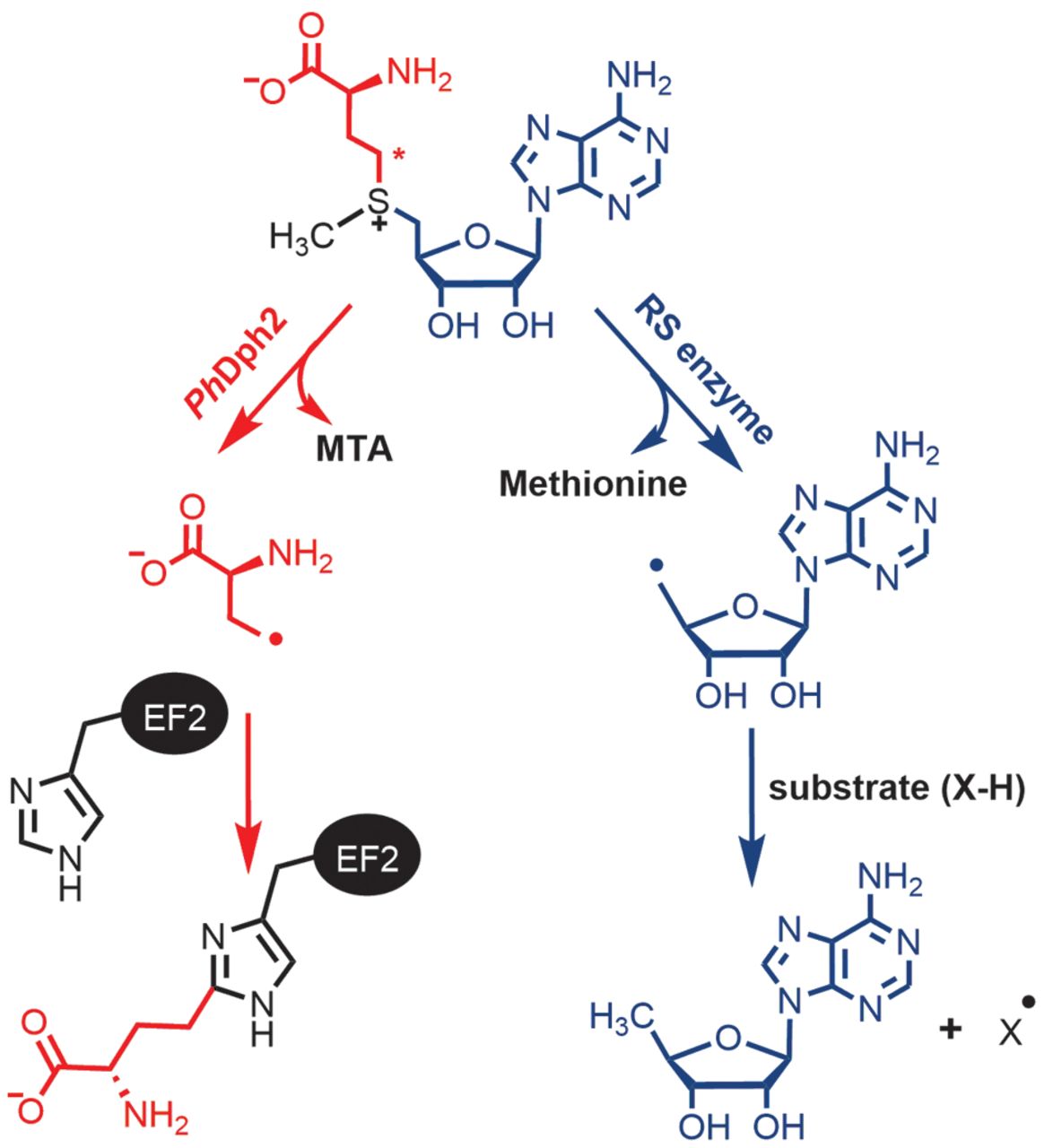
|
|
Organometallic and radical intermediates reveal mechanism of diphthamide biosynthesis
M. Dong, V. Kathiresan, M. K. Fenwick, A. T. Torelli, Y. Zhang, J. D. Caranto, B. Dzikovski, A. Sharma, K. M. Lancaster, J. H. Freed, S. E. Ealick, B. M. Hoffman, and H. Lin.
Science 359, 1247-1250 (2018)
Supporting Information
<doi: 10.1126/science.aao6595>
PMID:
29590073
PMCID:
PMC6066404
|
|
|
ABSTRACT: Diphthamide biosynthesis involves a carbon-carbon bond-forming reaction catalyzed by a radical S-adenosylmethionine (SAM) enzyme that cleaves a carbon-sulfur (C–S) bond in SAM to generate a 3-amino-3-carboxypropyl (ACP) radical. Using rapid freezing, we have captured an organometallic intermediate with an iron-carbon (Fe–C) bond between ACP and the enzyme's [4Fe-4S] cluster. In the presence of the substrate protein, elongation factor 2, this intermediate converts to an organic radical, formed by addition of the ACP radical to a histidine side chain. Crystal structures of archaeal diphthamide biosynthetic radical SAM enzymes reveal that the carbon of the SAM C–S bond being cleaved is positioned near the unique cluster Fe, able to react with the cluster. Our results explain how selective C–S bond cleavage is achieved in this radical SAM enzyme.
|
|
|
Protein dynamics in the solid-state from 2H NMR lineshape analysis. III. MOMD in the presence of Magic Angle Spinning
E. Meirovitch, Z. Liang, J. H. Freed.
Solid State Nucl. Magn. Reson. 89, 35-44 (2018)
<doi: 10.1016/j.ssnmr.2017.11.001>
PMID:
29208317
PMCID:
PMC5772661
|
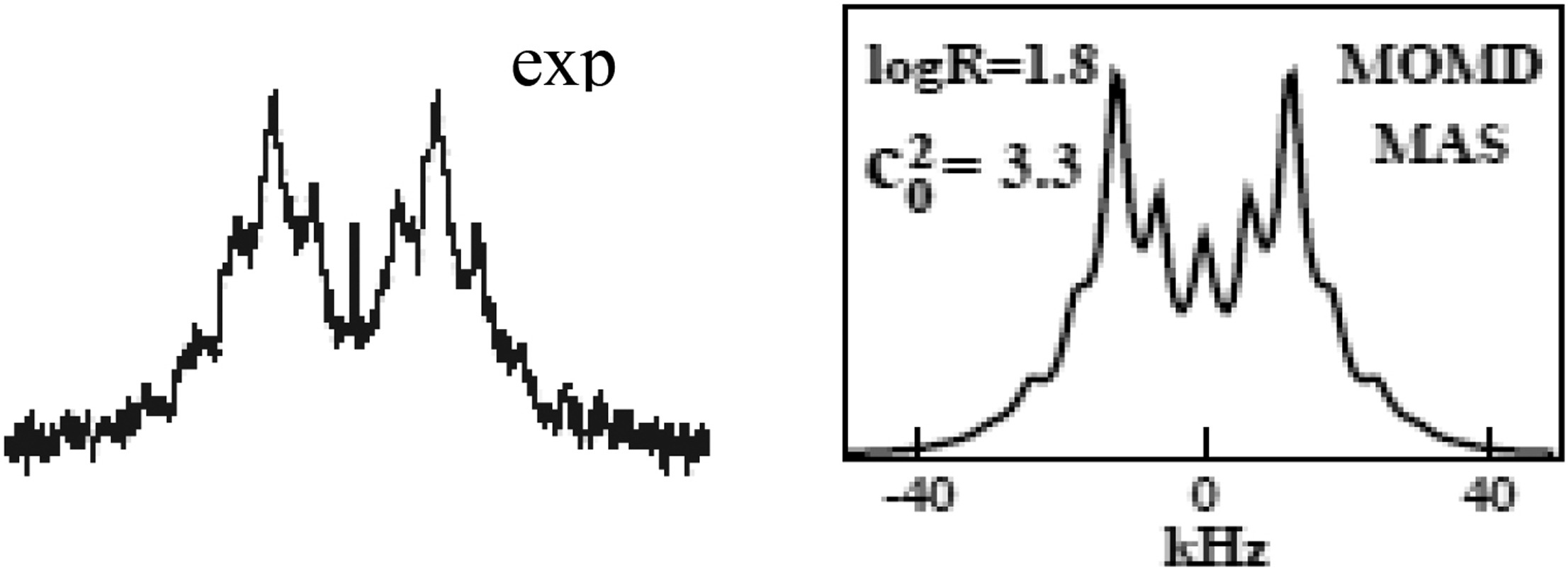
|
|
ABSTRACT: We report on a new approach to the analysis of dynamic NMR lineshapes from polycrystalline (i.e., macroscopically disordered) samples in the presence of Magic Angle Spinning (MAS). This is an application of the Stochastic Liouville Equation developed by Freed and co-workers for treating restricted (i.e., microscopically ordered) motions. The 2H nucleus in an internally-mobile C–CD3 moiety serves as a prototype probe. The acronym is 2H/MOMD/MAS, where MOMD stands for "microscopic-order-macroscopic-disorder." The key elements describing internal motions – their type, the local spatial restrictions, and related features of local geometry – are treated in MOMD generally, within their rigorous three-dimensional tensorial requirements. Based on this representation a single physically well-defined model of local motion has the capability of reproducing experimental spectra. There exist other methods for analyzing dynamic 2H/MAS spectra which advocate simple motional modes. Yet, to reproduce satisfactorily the experimental lineshapes, one has either to use unusual parameter values, or combine several simple motional modes. The multi-simple-mode reasoning assumes independence of the constituent modes, features ambiguity as different simple modes may be used, renders inter-system comparison difficult as the overall models differ, and makes possible model-improvement only by adding yet another simple mode, i.e., changing the overall model. 2H/MOMD/MAS is free of such limitations and inherently provides a clear physical interpretation. These features are illustrated. The advantage of 2H/MOMD/MAS in dealing with sensitive but hardly investigated slow-motional lineshapes is demonstrated by applying it to actual experimental data. The results differ from those obtained previously with a two-site exchange scheme that yielded unusual parameters.
|
|
|
The SARS-CoV Fusion Peptide Forms an Extended Bipartite Fusion Platform that Perturbs Membrane Order in a Calcium-Dependent Manner
A. L. Lai, J. K. Millet, S. Daniel, J. H. Freed, and G. R. Whittaker.
J. Mol. Biol. 429, 3875-3892 (2017)
<doi: 10.1016/j.jmb.2017.10.017>
PMID:
29056462
PMCID:
PMC5705393
|

|
|
ABSTRACT: Coronaviruses (CoVs) are a major infectious disease threat and include the pathogenic human pathogens of zoonotic origin: severe acute respiratory syndrome CoV (SARS-CoV) and Middle East respiratory syndrome CoV (MERS-CoV). Entry of CoVs into host cells is mediated by the viral spike (S) protein, which is structurally categorized as a class I viral fusion protein, within the same group as influenza virus and HIV. However, S proteins have two distinct cleavage sites that can be activated by a much wider range of proteases. The exact location of the CoV fusion peptide (FP) has been disputed. However, most evidence suggests that the domain immediately downstream of the S2′ cleavage site is the FP (amino acids 798–818 SFIEDLLFNKVTLADAGFMKQY for SARS-CoV, FP1). In our previous electron spin resonance spectroscopic studies, the membrane-ordering effect of influenza virus, HIV, and Dengue virus FPs has been consistently observed. In this study, we used this effect as a criterion to identify and characterize the bona fide SARS-CoV FP. Our results indicate that both FP1 and the region immediately downstream (amino acids 816–835 KQYGECLGDINARDLICAQKF, FP2) induce significant membrane ordering. Furthermore, their effects are calcium dependent, which is consistent with in vivo data showing that calcium is required for SARS-CoV S-mediated fusion. Isothermal titration calorimetry showed a direct interaction between calcium cations and both FPs. This Ca2+-dependency membrane ordering was not observed with influenza FP, indicating that the CoV FP exhibits a mechanistically different behavior. Membrane-ordering effects are greater and penetrate deeper into membranes when FP1 and FP2 act in a concerted manner, suggesting that they form an extended fusion "platform."
|
|
|
Dipolar Spectroscopy – Single-Resonance Methods
P. P. Borbat and J. H. Freed.
eMagRes 6 425-462 (2017)
<doi: 10.1002/9780470034590.emrstm1519>
PMID: [None - Book] PMCID:
[None - Book]
|
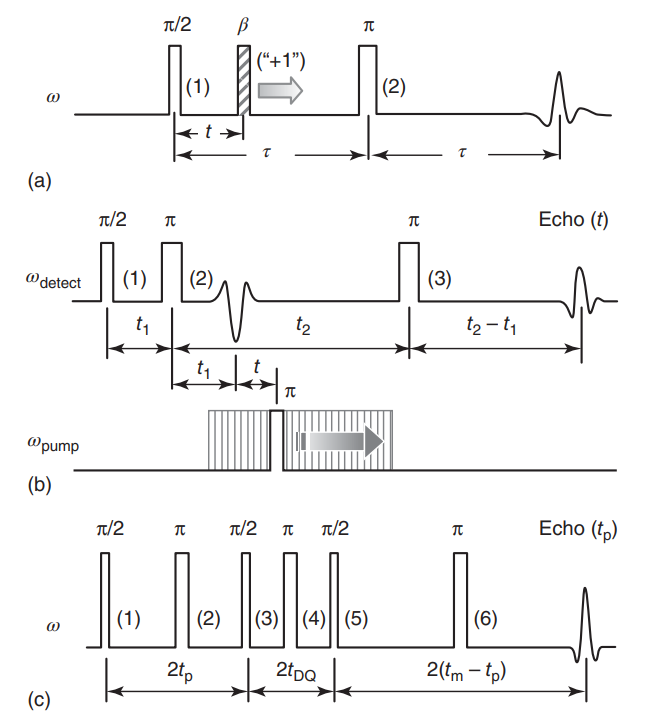
|
|
ABSTRACT: A range of powerful techniques based on pulse EPR spectroscopies is used extensively for measuring electron spin dipolar couplings between native or introduced paramagnetic reporters, from which accurate distances, and in certain cases molecular orientations, can be determined to aid in solving biomolecular structures. Notably, several versatile pulse EPR methods of high sensitivity were developed over the past two decades. The two main groups of pulse methods are represented by single- and double-resonance techniques, which together constitute pulse dipolar EPR spectroscopy (PDS). We describe the first group by outlining the basic principles and illustrate key techniques with several examples from the literature, as well as some newly produced for purposes of this chapter. Specifically, we describe double-quantum coherence (DQC) methods for robust selection of the dipolar coupling by filtering out undesired spin coherence pathways via phase cycling, and we also discuss single-resonance methods such as four- and five-pulse single-quantum coherence (SQC) methods, all of which benefit from maximizing the spectral coverage. The common features and conceptual differences between single- and double-resonance methods are discussed. In addition, the extensions of modern DQC and SQC methods to two-dimensional correlation spectroscopy for eliciting orientational information are included. Other types of 'single-frequency' PDS methods, not all of which are based on principles behind fully coherent single-resonance methods, are also discussed to show the diversity of PDS methods.
|
|
|
Singular Value Decomposition Method to Determine Distance Distributions in Pulsed Dipolar Electron Spin Resonance
M. Srivastava and J. H. Freed.
J. Phys. Chem. Lett. 8, 5648-5655 (2017)
Supporting Information
<doi: 10.1021/acs.jpclett.7b02379>
PMID:
29099190
PMCID:
PMC5708871
|
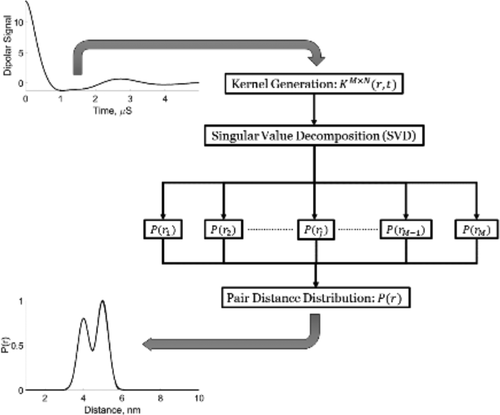
|
|
ABSTRACT: Regularization is often utilized to elicit the desired physical results from experimental data. The recent development of a denoising procedure yielding about 2 orders of magnitude in improvement in SNR obviates the need for regularization, which achieves a compromise between canceling effects of noise and obtaining an estimate of the desired physical results. We show how singular value decomposition (SVD) can be employed directly on the denoised data, using pulse dipolar electron spin resonance experiments as an example. Such experiments are useful in measuring distances and their distributions, P(r) between spin labels on proteins. In noise-free model cases exact results are obtained, but even a small amount of noise (e.g., SNR = 850 after denoising) corrupts the solution. We develop criteria that precisely determine an optimum approximate solution, which can readily be automated. This method is applicable to any signal that is currently processed with regularization of its SVD analysis.
|
|
|
Synthesis and Solution-Phase Characterization of Sulfonated Oligothioetheramides
J. S. Brown, Y. M. Acevedo, G. D. He, J. H. Freed, P. Clancy, and C. A. Alabi.
Macromolecules 50, 8731-8738 (2017)
Supporting Information
<doi: 10.1021/acs.macromol.7b01915>
PMID:
29386690
PMCID:
PMC5788177
|
|
|
ABSTRACT: Nature has long demonstrated the importance of chemical sequence to induce structure and tune physical interactions. Investigating macromolecular structure and dynamics is paramount to understand macromolecular binding and target recognition. To that end, we have synthesized and characterized flexible sulfonated oligothioetheramides (oligoTEAs) by variable temperature pulse field gradient (PFG) NMR, double electron–electron resonance (DEER), and molecular dynamics (MD) simulations to capture their room temperature structure and dynamics in water. We have examined the contributions of synthetic length (2–12mer), pendant group charge, and backbone hydrophobicity. We observe significant entropic collapse, driven in part by backbone hydrophobicity. Analysis of individual monomer contributions revealed larger changes due to the backbone compared to pendant groups. We also observe screening of intramolecular electrostatic repulsions. Finally, we comment on the combination of DEER and PFG NMR measurements via Stokes–Einstein–Sutherland diffusion theory. Overall, this sensitive characterization holds promise to enable de novo development of macromolecular structure and sequence–structure–function relationships with flexible, but biologically functional macromolecules.
|
|
|
Mechanistic Insight into the Photocontrolled Cationic Polymerization of Vinyl Ethers
Q. Michaudel, T. Chauviré, V. Kottisch, M. J. Supej, K. J. Stawiasz, L. Shen, W. R. Zipfel, H. D. Abruña, J. H. Freed, and B. P. Fors.
J. Am. Chem. Soc. 139, 15530-15538 (2017)
|
|
|
Mechanistic Insight into the Photocontrolled Cationic Polymerization of Vinyl Ethers
Q. Michaudel, T. Chauviré, V. Kottisch, M. J. Supej, K. J. Stawiasz, L. Shen, W. R. Zipfel, H. D. Abruña, J. H. Freed, and B. P. Fors.
J. Am. Chem. Soc. 139, 15530-15538 (2017)
Supporting Information
<doi: 10.1021/jacs.7b09539>
PMID:
28985061
PMCID:
PMC5806523
|
|
|
ABSTRACT: The mechanism of the recently reported photocontrolled cationic polymerization of vinyl ethers was investigated using a variety of catalysts and chain-transfer agents (CTAs) as well as diverse spectroscopic and electrochemical analytical techniques. Our study revealed a complex activation step characterized by one-electron oxidation of the CTA. This oxidation is followed by mesolytic cleavage of the resulting radical cation species, which leads to the generation of a reactive cation–this species initiates the polymerization of the vinyl ether monomer–and a dithiocarbamate radical that is likely in equilibrium with the corresponding thiuram disulfide dimer. Reversible addition–fragmentation type degenerative chain transfer contributes to the narrow dispersities and control over chain growth observed under these conditions. Finally, the deactivation step is contingent upon the oxidation of the reduced photocatalyst by the dithiocarbamate radical concomitant with the production of a dithiocarbamate anion that caps the polymer chain end. The fine-tuning of the electronic properties and redox potentials of the photocatalyst in both the excited and the ground states is necessary to obtain a photocontrolled system rather than simply a photoinitiated system. The elucidation of the elementary steps of this process will aid the design of new catalytic systems and their real-world applications.
|
|
|
Key features of an Hsp70 chaperone allosteric landscape revealed by ion-mobility native mass spectrometry and double electron-electron resonance
A. L. Lai, E. M. Clerico, M. E. Blackburn, N. A. Patel, C. V. Robinson, P. P. Borbat, J. H. Freed, and L. M. Gierasch.
J. Biol. Chem. 292, 8773-8785 (2017)
Supporting Information
<doi: 10.1074/jbc.M116.770404>
PMID:
28428246
PMCID:
PMC5448104
|
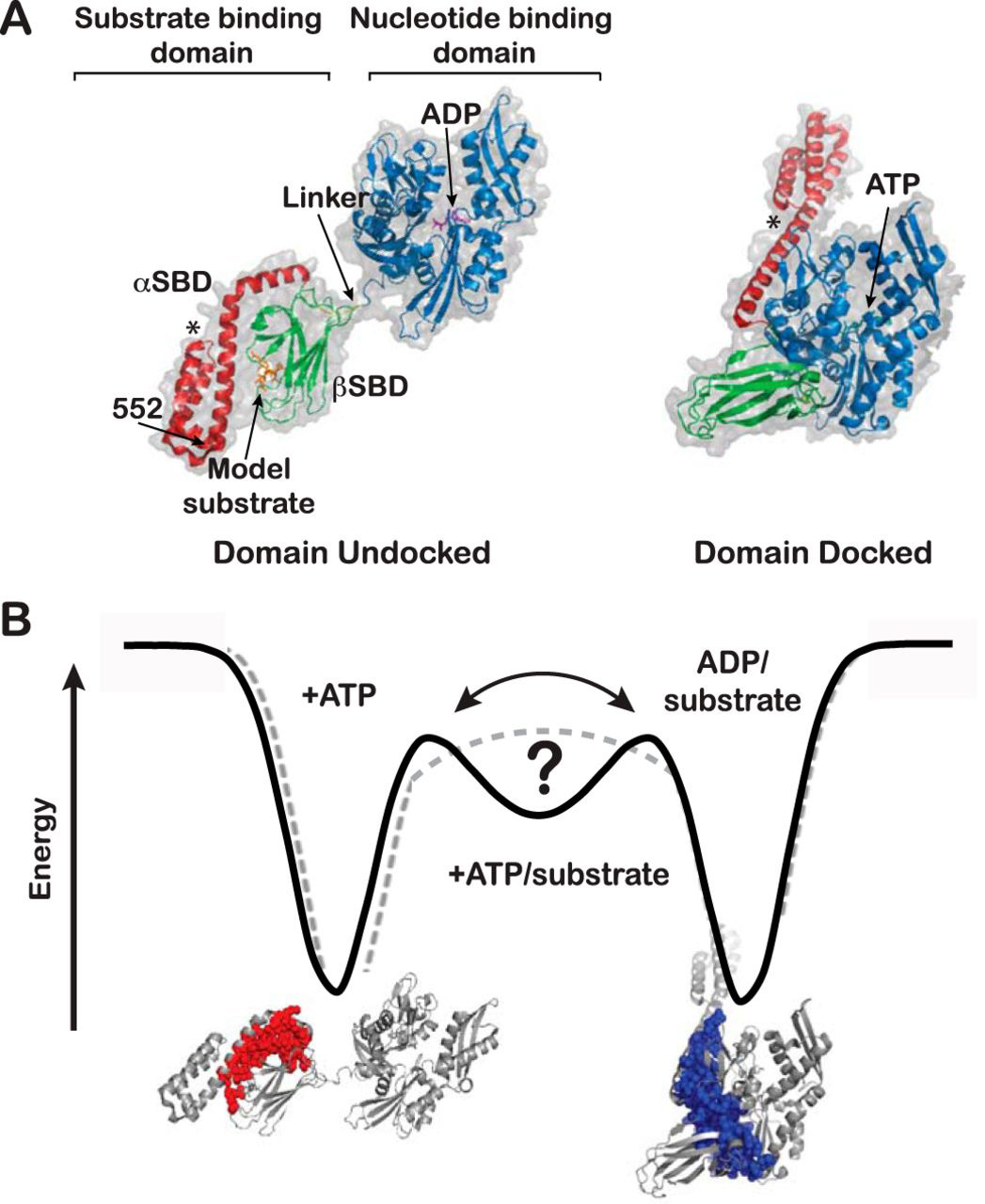
|
|
ABSTRACT: Proteins are dynamic entities that populate conformational ensembles, and most functions of proteins depend on their dynamic character. Allostery, in particular, relies on ligand-modulated shifts in these conformational ensembles. Hsp70s are allosteric molecular chaperones with conformational landscapes that involve large rearrangements of their two domains (viz. the nucleotide-binding domain and substrate-binding domain) in response to adenine nucleotides and substrates. However, it remains unclear how the Hsp70 conformational ensemble is populated at each point of the allosteric cycle and how ligands control these populations. We have mapped the conformational species present under different ligand-binding conditions throughout the allosteric cycle of the Escherichia coli Hsp70 DnaK by two complementary methods, ion-mobility mass spectrometry and double electron-electron resonance. Our results obtained under biologically relevant ligand-bound conditions confirm the current picture derived from NMR and crystallographic data of domain docking upon ATP binding and undocking in response to ADP and substrate. Additionally, we find that the helical lid of DnaK is a highly dynamic unit of the structure in all ligand-bound states. Importantly, we demonstrate that DnaK populates a partially docked state in the presence of ATP and substrate and that this state represents an energy minimum on the DnaK allosteric landscape. Because Hsp70s are emerging as potential drug targets for many diseases, fully mapping an allosteric landscape of a molecular chaperone like DnaK will facilitate the development of small molecules that modulate Hsp70 function via allosteric mechanisms.
|
|
|
Unique Structural Features of Membrane-Bound C-Terminal Domain Motifs Modulate Complexin Inhibitory Function
D. Snead, A. L. Lai, R. T. Wragg, D. A. Parisotto, T. F. Ramlall, J. S. Dittman, J. H. Freed, and D. Eliezer.
Front. Mol. Neurosci. 10, 154 (2017)
Supporting Information
<doi: 10.3389/fnmol.2017.00154>
PMID:
28596722
PMCID:
PMC5442187
|

|
|
ABSTRACT: Complexin is a small soluble presynaptic protein that interacts with neuronal SNARE proteins in order to regulate synaptic vesicle exocytosis. While the SNARE-binding central helix of complexin is required for both the inhibition of spontaneous fusion and the facilitation of synchronous fusion, the disordered C-terminal domain (CTD) of complexin is specifically required for its inhibitory function. The CTD of worm complexin binds to membranes via two distinct motifs, one of which undergoes a membrane curvature dependent structural transition that is required for efficient inhibition of neurotransmitter release, but the conformations of the membrane-bound motifs remain poorly characterized. Visualizing these conformations is required to clarify the mechanisms by which complexin membrane interactions regulate its function. Here, we employ optical and magnetic resonance spectroscopy to precisely define the boundaries of the two CTD membrane-binding motifs and to characterize their conformations. We show that the curvature dependent amphipathic helical motif features an irregular element of helical structure, likely a pi-bulge, and that this feature is important for complexin inhibitory function in vivo.
|
|
|
Stability and Conformation of a Chemoreceptor HAMP Domain Chimera Correlates with Signaling Properties
N. Sukomon, J. Widom, P. P. Borbat, J. H. Freed, and B. R. Crane.
Biophys. J. 112, 1383-1395 (2017)
Supporting Information
<doi: 10.1016/j.bpj.2017.02.037>
PMID:
28402881
PMCID:
PMC5390053
|
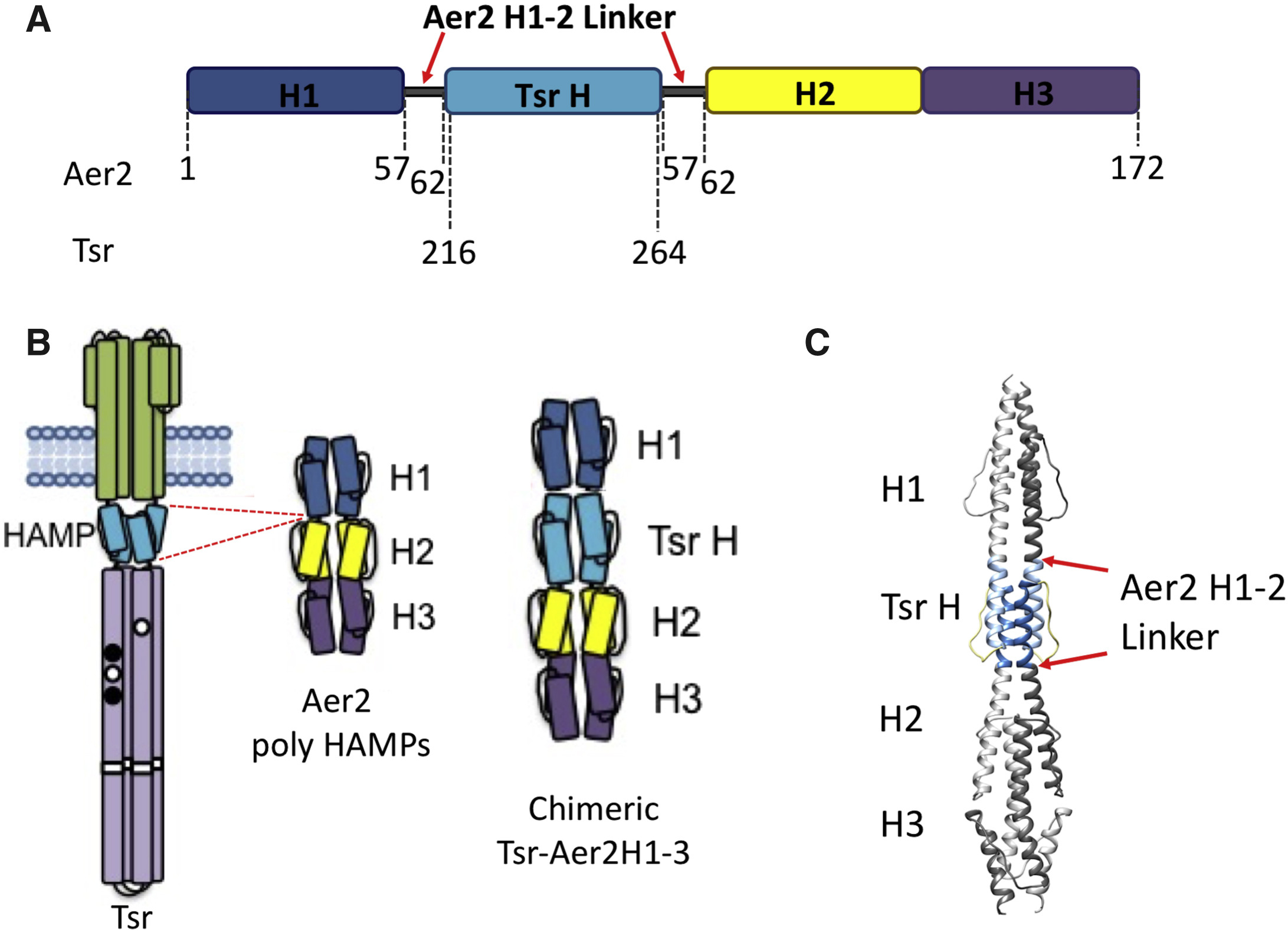
|
|
ABSTRACT: HAMP domains are dimeric, four-helix bundles that transduce conformational signals in bacterial receptors. Genetic studies of the Escherichia coli serine receptor (Tsr) provide an opportunity to understand HAMP conformational behavior in terms of functional output. To increase its stability, the Tsr HAMP domain was spliced into a poly-HAMP unit from the Pseudomonas aeruginosa Aer2 receptor. Within the chimera, the Tsr HAMP undergoes a thermal melting transition at a temperature much lower than that of the Aer2 HAMP domains. Pulse-dipolar electron spin resonance spectroscopy and site-specific spin-labeling confirm that the Tsr HAMP maintains a four-helix bundle. Pulse-dipolar electron spin resonance spectroscopy was also used to study three well-characterized HAMP mutational phenotypes: those that cause flagella rotation that is counterclockwise (CCW) A and kinase-off; CCW B and also kinase-off; and, clockwise (CW) and kinase-on. Conformational properties of the three HAMP variants support a biphasic model of dynamic bundle stability, but also indicate distinct conformational changes within the helix bundle. Functional kinase-on (CW) and kinase-off (CCW A) states differ by concerted changes in the positions of spin-label sites at the base of the bundle. Opposite shifts in the subunit separation distances of neighboring residues at the C-termini of the α1 and α2 helices are consistent with a helix scissors motion or a gearbox rotational model of HAMP activation. In the drastic kinase-off lesion of CCW B, the α1 helices unfold and the α2 helices form a tight two-helix coiled-coil. The substitution of a critical residue in the Tsr N-terminal linker or control cable reduces conformational heterogeneity at the N-terminus of α1 but does not affect structure at the C-terminus of α2. Overall, the data suggest that transitions from on- to off-states involve decreased motional amplitudes of the Tsr HAMP coupled with helix rotations and movements toward a two-helix packing mode.
|
|
|
Substrate-Dependent Cleavage Site Selection by Unconventional Radical S-Adenosylmethionine Enzymes in Diphthamide Biosynthesis
M. Dong, M. Horitani, B. Dzikovski, J. H. Freed, S. E. Ealick, B. M. Hoffman, and H. Lin.
J. Am. Chem. Soc. 139, 5680-5683 (2017)
Supporting Information
<doi: 10.1021/jacs.7b01712>
PMID:
28383907
PMCID:
PMC5664936
|
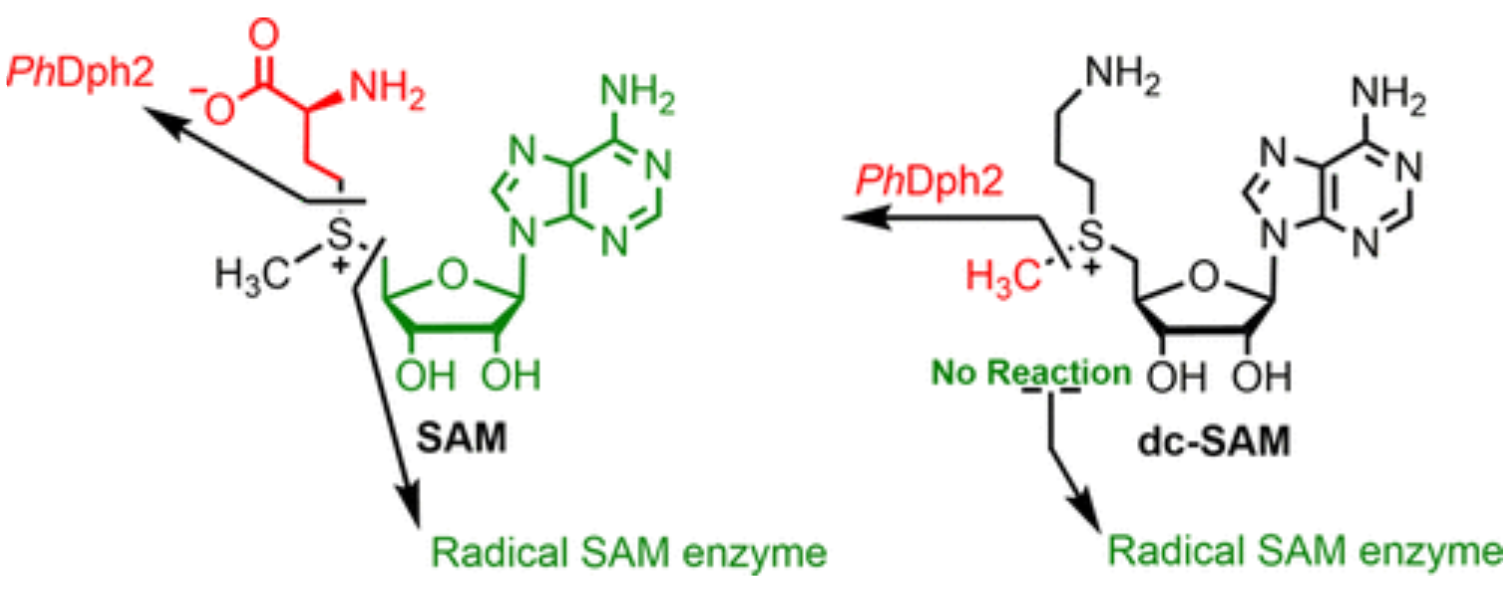
|
|
ABSTRACT: S-Adenosylmethionine (SAM) has a sulfonium ion with three distinct C-S bonds. Conventional radical SAM enzymes use a [4Fe-4S] cluster to cleave homolytically the C5′,adenosine-S bond of SAM to generate a 5′-deoxyadenosyl radical, which catalyzes various downstream chemical reactions. Radical SAM enzymes involved in diphthamide biosynthesis, such as Pyrococcus horikoshii Dph2 (PhDph2) and yeast Dph1-Dph2 instead cleave the Cγ,Met-S bond of methionine to generate a 3-amino-3-carboxylpropyl radical. We here show radical SAM enzymes can be tuned to cleave the third C-S bond to the sulfonium sulfur by changing the structure of SAM. With a decarboxyl SAM analogue (dc-SAM), PhDph2 cleaves the Cmethyl-S bond, forming 5′-deoxy-5′-(3-aminopropylthio) adenosine (dAPTA, 1). The methyl cleavage activity, like the cleavage of the other two C-S bonds, is dependent on the presence of a [4Fe-4S]+ cluster. Electron-nuclear double resonance and mass spectroscopy data suggests that mechanistically one of the S atoms in the [4Fe-4S] cluster captures the methyl group from dc-SAM, forming a distinct EPR-active intermediate, which can transfer the methyl group to nucleophiles such as dithiothreitol. This reveals the [4Fe-4S] cluster in a radical SAM enzyme can be tuned to cleave any one of the three bonds to the sulfonium sulfur of SAM or analogues, and is the first demonstration a radical SAM enzyme could switch from an Fe-based one electron transfer reaction to a S-based two electron transfer reaction in a substrate-dependent manner. This study provides an illustration of the versatile reactivity of Fe-S clusters.
|
|
|
Signature of an aggregation-prone conformation of tau
N. A. Eschmann, E. R. Georgieva, P. Ganguly, P. P. Borbat, M. D. Rappaport, Y. Akdogan, J. H. Freed, J. -E. Shea, and S. Han.
Sci. Rep. 7, 44739 (2017)
Supporting Information
<doi: 10.1038/srep44739>
PMID:
28303942
PMCID:
PMC5356194
|
|
|
ABSTRACT: The self-assembly of the microtubule associated tau protein into fibrillar cell inclusions is linked to a number of devastating neurodegenerative disorders collectively known as tauopathies. The mechanism by which tau self-assembles into pathological entities is a matter of much debate, largely due to the lack of direct experimental insights into the earliest stages of aggregation. We present pulsed double electron-electron resonance measurements of two key fibril-forming regions of tau, PHF6 and PHF6*, in transient as aggregation happens. By monitoring the end-to-end distance distribution of these segments as a function of aggregation time, we show that the PHF6(*) regions dramatically extend to distances commensurate with extended β-strand structures within the earliest stages of aggregation, well before fibril formation. Combined with simulations, our experiments show that the extended β-strand conformational state of PHF6(*) is readily populated under aggregating conditions, constituting a defining signature of aggregation-prone tau, and as such, a possible target for therapeutic interventions.
|
|
|
A New Wavelet Denoising Method for Experimental Time-Domain Signals: Pulsed Dipolar Electron Spin Resonance
M. Srivastava, E. R. Georgieva, and J. H. Freed.
J. Phys. Chem. A 121 2452-2465 (2017)
Supporting Information
<doi: 10.1021/acs.jpca.7b00183>
PMID:
28257206
PMCID:
PMC5553325
|
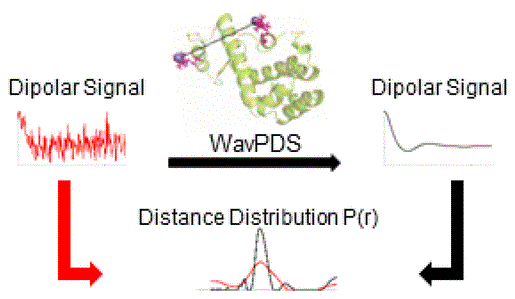
|
|
ABSTRACT: We adapt a new wavelet-transform-based method of denoising experimental signals to pulse-dipolar electron-spin resonance spectroscopy (PDS). We show that signal averaging times of the time-domain signals can be reduced by as much as 2 orders of magnitude, while retaining the fidelity of the underlying signals, in comparison with noiseless reference signals. We have achieved excellent signal recovery when the initial noisy signal has an SNR ≳ 3. This approach is robust and is expected to be applicable to other time-domain spectroscopies. In PDS, these time-domain signals representing the dipolar interaction between two electron spin labels are converted into their distance distribution functions P(r), usually by regularization methods such as Tikhonov regularization. The significant improvements achieved by using denoised signals for this regularization are described. We show that they yield P(r)'s with more accurate detail and yield clearer separations of respective distances, which is especially important when the P(r)'s are complex. Also, longer distance P(r)'s, requiring longer dipolar evolution times, become accessible after denoising. In comparison to standard wavelet denoising approaches, it is clearly shown that the new method (WavPDS) is superior.
|
|
|
Structure-Function Studies Link Class II Viral Fusogens with the Ancestral Gamete Fusion Protein HAP2
J. F. Pinello, A. L. Lai, J. K. Millet, D. Cassidy-Hanley, J. H. Freed, and T. G. Clark.
Curr. Biol. 27, 651-660 (2017)
Supporting Information
<doi: 10.1016/j.cub.2017.01.049>
PMID:
28238660
PMCID:
PMC5393271
|
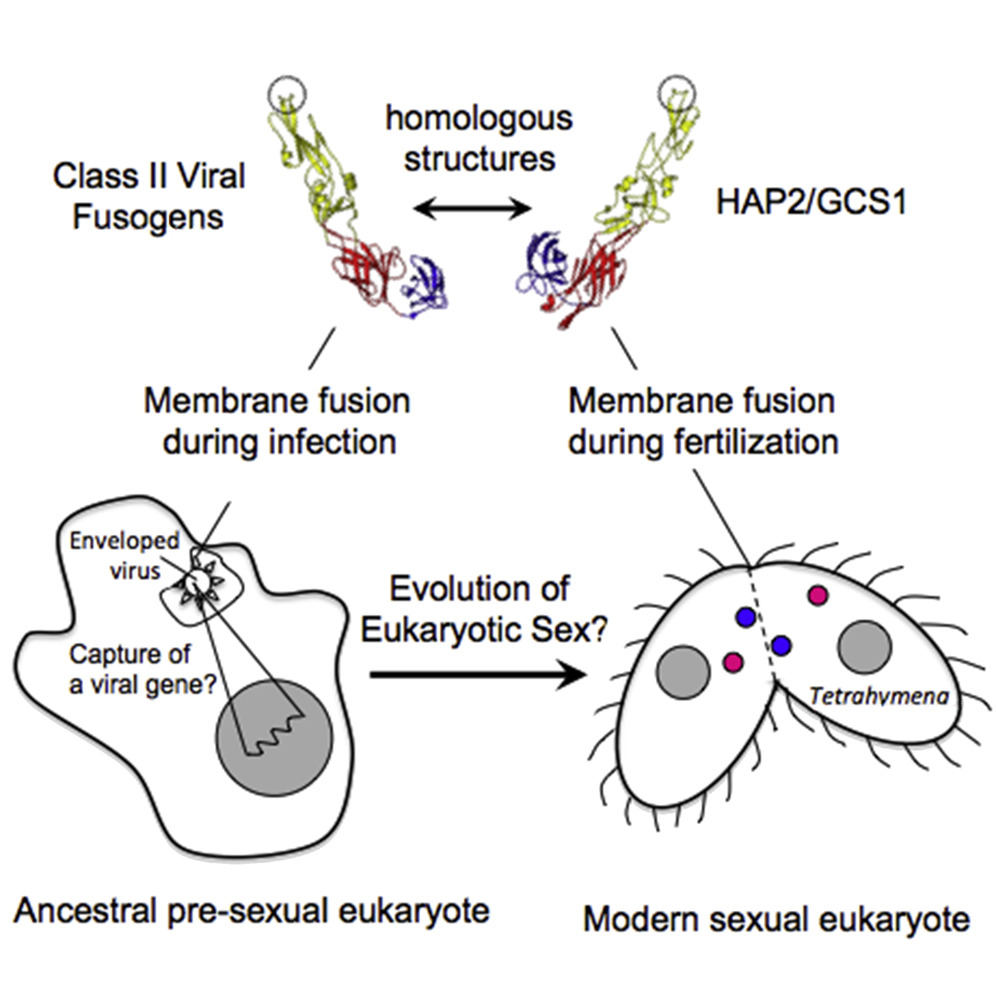
|
|
ABSTRACT: The conserved transmembrane protein, HAP2/GCS1, has been linked to fertility in a wide range of taxa and is hypothesized to be an ancient gamete fusogen. Using template-based structural homology modeling, we now show that the ectodomain of HAP2 orthologs from Tetrahymena thermophila and other species adopt a protein fold remarkably similar to the dengue virus E glycoprotein and related class II viral fusogens. To test the functional significance of this predicted structure, we developed a flow-cytometry-based assay that measures cytosolic exchange across the conjugation junction to rapidly probe the effects of HAP2 mutations in the Tetrahymena system. Using this assay, alterations to a region in and around a predicted "fusion loop" in T. thermophila HAP2 were found to abrogate membrane pore formation in mating cells. Consistent with this, a synthetic peptide corresponding to the HAP2 fusion loop was found to interact directly with model membranes in a variety of biophysical assays. These results raise interesting questions regarding the evolutionary relationships of class II membrane fusogens and harken back to a long-held argument that eukaryotic sex arose as the byproduct of selection for the horizontal transfer of a "selfish" genetic element from cell to cell via membrane fusion.
|
|
|
Bacterial Energy Sensor Aer Modulates the Activity of the Chemotaxis Kinase CheA Based on the Redox State of the Flavin Cofactor
D. Samanta, J. Widom, P. P. Borbat, J. H. Freed, and B. R. Crane.
J. Biol. Chem. 291, 25809-25814 (2016)
<doi: 10.1074/jbc.C116.757492>
PMID:
27803157
PMCID:
PMC5207056
|
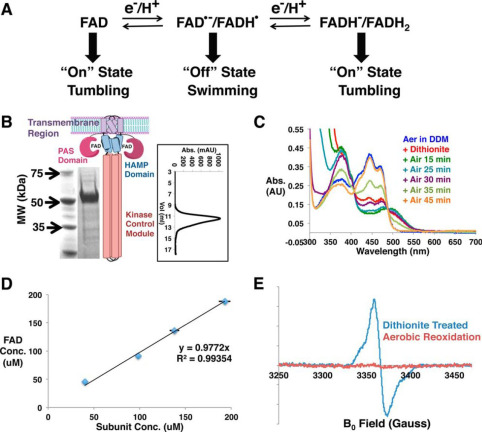
|
|
ABSTRACT: Flagellated bacteria modulate their swimming behavior in response to environmental cues through the CheA/CheY signaling pathway. In addition to responding to external chemicals, bacteria also monitor internal conditions that reflect the availability of oxygen, light, and reducing equivalents, in a process termed "energy taxis." In Escherichia coli, the transmembrane receptor Aer is the primary energy sensor for motility. Genetic and physiological data suggest that Aer monitors the electron transport chain through the redox state of its FAD cofactor. However, direct biochemical data correlating FAD redox chemistry with CheA kinase activity have been lacking. Here, we test this hypothesis via functional reconstitution of Aer into nanodiscs. As purified, Aer contains fully oxidized FAD, which can be chemically reduced to the anionic semiquinone (ASQ). Oxidized Aer activates CheA, whereas ASQ Aer reversibly inhibits CheA. Under these conditions, Aer cannot be further reduced to the hydroquinone, in contrast to the proposed Aer signaling model. Pulse ESR spectroscopy of the ASQ corroborates a potential mechanism for signaling in that the resulting distance between the two flavin-binding PAS (Per-Arnt-Sim) domains implies that they tightly sandwich the signal-transducing HAMP domain in the kinase-off state. Aer appears to follow oligomerization patterns observed for related chemoreceptors, as higher loading of Aer dimers into nanodiscs increases kinase activity. These results provide a new methodological platform to study Aer function along with new mechanistic details into its signal transduction process.
|
|
|
Design of Hydrated Porphyrin-Phospholipid Bilayers with Enhanced Magnetic Resonance Contrast
S. Shao, T. N. Do, A. Razi, U. Chitgupi, J. Geng, R. J. Alsop, B. G. Dzikovski, M. C. Rheinstädter, J. Ortega, M. Karttunen, J. A. Spernyak, and J. F. Lovell.
Small 13, 1602505 (2017).
|

|
|
Design of Hydrated Porphyrin-Phospholipid Bilayers with Enhanced Magnetic Resonance Contrast
S. Shao, T. N. Do, A. Razi, U. Chitgupi, J. Geng, R. J. Alsop, B. G. Dzikovski, M. C. Rheinstädter, J. Ortega, M. Karttunen, J. A. Spernyak, and J. F. Lovell.
Small 13, 1602505 (2017).
Supporting Information
<doi: 10.1002/smll.201602505>
PMID:
27739249
PMCID:
PMC5209247
|
|
|
ABSTRACT: Computer simulations are used to design more hydrated bilayers, formed from amine-modified porphyrin-phospholipids (PoPs). Experiments confirm that the new constructs give rise to bilayers with greater water content. When chelated with manganese, amine-modified PoPs provide improved contrast for magnetic resonance and are safely used for imaging in vivo.
|
|
|
Constraints on the Radical Cation Center of Cytochrome c Peroxidase for Electron Transfer from Cytochrome c
T. M. Payne, E. F. Yee, B. Dzikovski, and B. R. Crane.
Biochemistry 55, 4807-4822 (2016).
<doi: 10.1021/acs.biochem.6b00262>
PMID:
27499202
PMCID:
PMC5689384
|

|
|
ABSTRACT: The tryptophan 191 cation radical of cytochrome c peroxidase (CcP) compound I (Cpd I) mediates long-range electron transfer (ET) to cytochrome c (Cc). Here we test the effects of chemical substitution at position 191. CcP W191Y forms a stable tyrosyl radical upon reaction with peroxide and produces spectral properties similar to those of Cpd I but has low reactivity toward reduced Cc. CcP W191G and W191F variants also have low activity, as do redox ligands that bind within the W191G cavity. Crystal structures of complexes between Cc and CcP W191X (X = Y, F, or G), as well as W191G with four bound ligands reveal similar 1:1 association modes and heme pocket conformations. The ligands display structural disorder in the pocket and do not hydrogen bond to Asp235, as does Trp191. Well-ordered Tyr191 directs its hydroxyl group toward the porphyrin ring, with no basic residue in the range of interaction. CcP W191X (X = Y, F, or G) variants substituted with zinc-porphyrin (ZnP) undergo photoinduced ET with Cc(III). Their slow charge recombination kinetics that result from loss of the radical center allow resolution of difference spectra for the charge-separated state [ZnP+, Cc(II)]. The change from a phenyl moiety at position 191 in W191F to a water-filled cavity in W191G produces effects on ET rates much weaker than the effects of the change from Trp to Phe. Low net reactivity of W191Y toward Cc(II) derives either from the inability of ZnP+ or the Fe-CcP ferryl to oxidize Tyr or from the low potential of the resulting neutral Tyr radical.
|
|
|
Organometallic Complex Formed by an Unconventional Radical S-Adenosylmethionine Enzyme
M. Dong, M. Horitani, B. Dzikovski, M. -E. Pandelia, C. Krebs, J. H. Freed, B. M. Hoffman, and H. Lin.
J. Am. Chem. Soc. 138, 9755-9758 (2016)
Supporting Information
<doi: 10.1021/jacs.6b04155>
PMID:
27465315
PMCID:
PMC5068486
|

|
|
ABSTRACT: Pyrococcus horikoshii Dph2 (PhDph2) is an unusual radical S-adenosylmethionine (SAM) enzyme involved in the first step of diphthamide biosynthesis. It catalyzes the reaction by cleaving SAM to generate a 3-amino-3-carboxypropyl (ACP) radical. To probe the reaction mechanism, we synthesized a SAM analogue (SAMCA), in which the ACP group of SAM is replaced with a 3-carboxyallyl group. SAMCA is cleaved by PhDph2, yielding a paramagnetic (S = 1/2) species, which is assigned to a complex formed between the reaction product, α-sulfinyl-3-butenoic acid, and the [4Fe-4S] cluster. Electron–nuclear double resonance (ENDOR) measurements with 13C and 2H isotopically labeled SAMCA support a π-complex between the C=C double bond of α-sulfinyl-3-butenoic acid and the unique iron of the [4Fe-4S] cluster. This is the first example of a radical SAM-related [4Fe-4S]+ cluster forming an organometallic complex with an alkene, shedding additional light on the mechanism of PhDph2 and expanding our current notions for the reactivity of [4Fe-4S] clusters in radical SAM enzymes.
|
|
|
Conformational Response of Influenza A M2 Transmembrane Domain to Amantadine Drug Binding at Low pH (pH 5.5)
E. R. Georgieva, P. P. Borbat, K. Grushin, S. Stoilova-McPhie, N. J. Kulkarni, Z. Liang, and J. H. Freed.
Front. Physiol. 7, 317 (2016)
Supporting Information
<doi: 10.3389/fphys.2016.00317>
PMID:
27524969
PMCID:
PMC4965473
|
|
|
ABSTRACT: The M2 protein from influenza A plays important roles in its viral cycle. It contains a single transmembrane helix, which oligomerizes into a homotetrameric proton channel that conducts in the low-pH environment of the host-cell endosome and Golgi apparatus, leading to virion uncoating at an early stage of infection. We studied conformational rearrangements that occur in the M2 core transmembrane domain residing on the lipid bilayer, flanked by juxtamembrane residues (M2TMD21–49 fragment), upon its interaction with amantadine drug at pH 5.5 when M2 is conductive. We also tested the role of specific mutation and lipid chain length. Electron spin resonance (ESR) spectroscopy and electron microscopy were applied to M2TMD21–49, labeled at the residue L46C with either nitroxide spin-label or Nanogold® reagent, respectively. Electron microscopy confirmed that M2TMD21–49 reconstituted into DOPC/POPS at 1:10,000 peptide-to-lipid molar ratio (P/L) either with or without amantadine, is an admixture of monomers, dimers, and tetramers, confirming our model based on a dimer intermediate in the assembly of M2TMD21–49. As reported by double electron-electron resonance (DEER), in DOPC/POPS membranes amantadine shifts oligomer equilibrium to favor tetramers, as evidenced by an increase in DEER modulation depth for P/L's ranging from 1:18,000 to 1:160. Furthermore, amantadine binding shortens the inter-spin distances (for nitroxide labels) by 5–8 Å, indicating drug induced channel closure on the C-terminal side. No such effect was observed for the thinner membrane of DLPC/DLPS, emphasizing the role of bilayer thickness. The analysis of continuous wave (cw) ESR spectra of spin-labeled L46C residue provides additional support to a more compact helix bundle in amantadine-bound M2TMD21–49 through increased motional ordering. In contrast to wild-type M2TMD21–49, the amantadine-bound form does not exhibit noticeable conformational changes in the case of G34A mutation found in certain drug-resistant influenza strains. Thus, the inhibited M2TMD21–49 channel is a stable tetramer with a closed C-terminal exit pore. This work is aimed at contributing to the development of structure-based anti-influenza pharmaceuticals.
|
|
|
A New Wavelet Denoising Method for Selecting Decomposition Levels and Noise Thresholds
M. Srivastava, C. L. Anderson, and J. H. Freed.
IEEE Access 4 3862-3877 (2016)
<doi: 10.1109/ACCESS.2016.2587581>
PMID:
27795877
PMCID:
PMC5079185
|
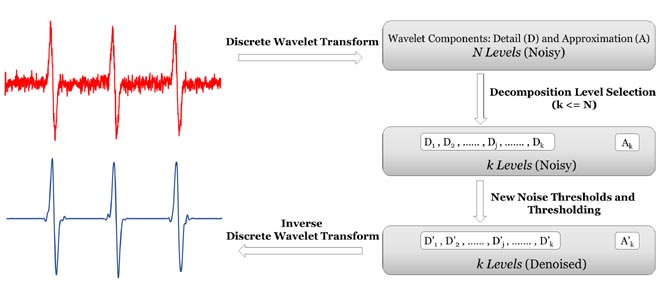
|
|
ABSTRACT: A new method is presented to denoise 1-D experimental signals using wavelet transforms. Although the state-of-the-art wavelet denoising methods perform better than other denoising methods, they are not very effective for experimental signals. Unlike images and other signals, experimental signals in chemical and biophysical applications, for example, are less tolerant to signal distortion and under-denoising caused by the standard wavelet denoising methods. The new method: 1) provides a method to select the number of decomposition levels to denoise; 2) uses a new formula to calculate noise thresholds that does not require noise estimation; 3) uses separate noise thresholds for positive and negative wavelet coefficients; 4) applies denoising to the approximation component; and 5) allows the flexibility to adjust the noise thresholds. The new method is applied to continuous wave electron spin resonance spectra and it is found that it increases the signal-to-noise ratio (SNR) by more than 32 dB without distorting the signal, whereas standard denoising methods improve the SNR by less than 10 dB and with some distortion. In addition, its computation time is more than six times faster.
|
|
|
Local Ordering at Mobile Sites in Proteins from Nuclear Magnetic Resonance Relaxation: the Role of Site Symmetry
O. Tchaicheeyan, J. H. Freed, and E. Meirovitch.
J. Phys. Chem. B 120 2886-2898 (2016)
Supporting Information
<doi: 10.1021/acs.jpcb.6b00524>
PMID:
26938937
PMCID:
PMC4837759
|
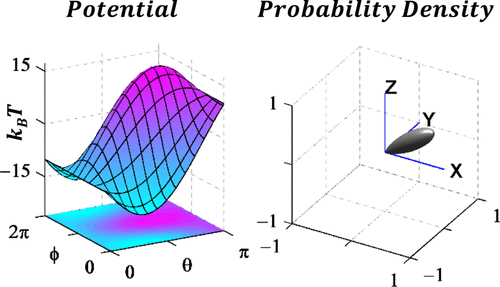
|
|
ABSTRACT: Restricted motions in proteins (e.g., N–H bond dynamics) are studied effectively with NMR. By analogy with restricted motions in liquid crystals (LC), the local ordering has in the past been primarily represented by potentials comprising the L = 2, |K| = 0, 2 spherical harmonics. However, probes dissolved in LCs experience nonpolar ordering, often referred to as alignment, while protein-anchored probes experience polar ordering, often referred to as orientation. In this study we investigate the role of local (site) symmetry in the context of the polarity of the local ordering. We find that potentials comprising the L = 1, |K| = 0, 1 spherical harmonics represent adequately polar ordering. It is useful to characterize potential symmetry in terms of the irreducible representations of D2h point group, which is already implicit in the definition of the rotational diffusion tensor. Thus, the relevant rhombic L = 1 potentials have B1u and B3u symmetry whereas the relevant rhombic L = 2 potentials have Ag symmetry. A comprehensive scheme where local potentials and corresponding probability density functions (PDFs) are represented in Cartesian and spherical coordinates clarifies how they are affected by polar and nonpolar ordering. The Cartesian coordinates are chosen so that the principal axis of polar axial PDF is pointing along the z-axis, whereas the principal axis of the nonpolar axial PDF is pointing along ±z. Two-term axial potentials with 1 ≤ L ≤ 3 exhibit substantial diversity; they are expected to be useful in NMR-relaxation-data-fitting. It is shown how potential coefficients are reflected in the experimental order parameters. The comprehensive scheme representing local potentials and PDFs is exemplified for the L = 2 case using experimental data from 15N-labeled plexin-B1 and thioredoxin, 2H-, and 13C-labeled benzenehexa-n-alkanoates, and nitroxide-labeled T4 lysozyme. Future prospects for improved ordering analysis based on combined atomistic and mesoscopic approaches are delineated.
|
|
|
Study of paramagnetic defect centers in as-grown and annealed TiO2 anatase and rutile nanoparticles by a variable-temperature X-band and high-frequency (236 GHz) EPR
S. K. Misra, S. I. Andronenko, D. Tipikin, J. H. Freed, V. Somani, and O. Prakash.
J. Magn. Magn. Mater. 401, 495-505 (2016)
<doi: 10.1016/j.jmmm.2015.10.072>
PMID:
27041794
PMCID:
PMC4815036
|

|
|
ABSTRACT: Detailed EPR investigations on as-grown and annealed TiO2 nanoparticles in the anatase and rutile phases were carried out at X-band (9.6 GHz) at 77, 120–300 K and at 236 GHz at 292 K. The analysis of EPR data for as-grown and annealed anatase and rutile samples revealed the presence of several paramagnetic centers: Ti3+, O−, adsorbed oxygen (O2−) and oxygen vacancies. On the other hand, in as-grown rutile samples, there were observed EPR lines due to adsorbed oxygen (O2−) and the Fe3+ ions in both Ti4+ substitutional positions, with and without coupling to an oxygen vacancy in the near neighborhood. Anatase nanoparticles were completely converted to rutile phase when annealed at 1000° C, exhibiting EPR spectra similar to those exhibited by the as-grown rutile nanoparticles. The high-frequency (236 GHz) EPR data on anatase and rutile samples, recorded in the region about g=2.0 exhibit resolved EPR lines, due to O− and O2− ions enabling determination of their g-values with higher precision, as well as observation of hyperfine sextets due to Mn2+ and Mn4+ ions in anatase.
|
|
|
Structural Basis for Activation, Assembly and Membrane Binding of ESCRT-III Snf7 Filaments
S. Tang, W. M. Henne, P. P. Borbat, N. J. Buchkovich, J. H. Freed, Y. Mao, J. C. Fromme, and S. D. Emr.
eLife 4 e12548 (2015)
Supporting Information
<doi: 10.7554/eLife.12548>
PMID:
26670543
PMCID:
PMC4720517
|
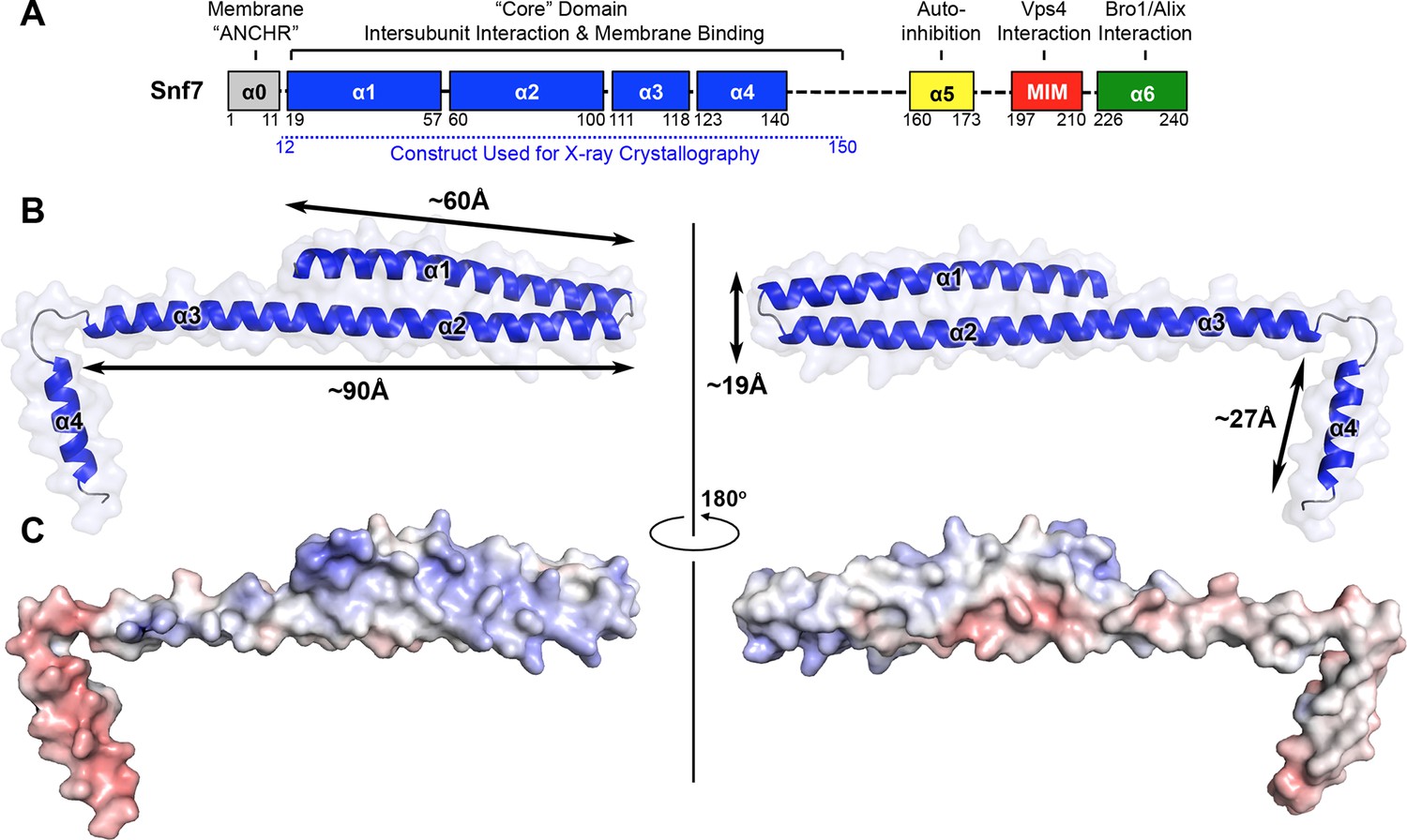
|
|
ABSTRACT: The endosomal sorting complexes required for transport (ESCRTs) constitute hetero-oligomeric machines that catalyze multiple topologically similar membrane-remodeling processes. Although ESCRT-III subunits polymerize into spirals, how individual ESCRT-III subunits are activated and assembled together into a membrane-deforming filament remains unknown. Here, we determine X-ray crystal structures of the most abundant ESCRT-III subunit Snf7 in its active conformation. Using pulsed dipolar electron spin resonance spectroscopy (PDS), we show that Snf7 activation requires a prominent conformational rearrangement to expose protein-membrane and protein-protein interfaces. This promotes the assembly of Snf7 arrays with ˜30 Å periodicity into a membrane-sculpting filament. Using a combination of biochemical and genetic approaches, both in vitro and in vivo, we demonstrate that mutations on these protein interfaces halt Snf7 assembly and block ESCRT function. The architecture of the activated and membrane-bound Snf7 polymer provides crucial insights into the spatially unique ESCRT-III-mediated membrane remodeling.
|
|
|
The Interaction between Influenza HA Fusion Peptide and Transmembrane Domain Affects Membrane Structure
A.L. Lai and J.H. Freed
Biophys. J. 109, 2523-2536 (2015)
Supporting Information
<doi: 10.1016/j.bpj.2015.10.044>
PMID:
26682811
PMCID:
PMC4699882
|

|
|
ABSTRACT: Viral glycoproteins, such as influenza hemagglutinin (HA) and human immunodeficiency virus gp41, are anchored by a single helical segment transmembrane domain (TMD) on the viral envelope membrane. The fusion peptides (FP) of the glycoproteins insert into the host membrane and initiate membrane fusion. Our previous study showed that the FP or TMD alone perturbs membrane structure. Interaction between the influenza HA FP and TMD has previously been shown, but its role is unclear. We used PC spin labels dipalmitoylphospatidyl-tempo-choline (on the headgroup), 5PC and 14PC (5-C and 14-C positions on the acyl chain) to detect the combined effect of FP-TMD interaction by titrating HA FP to TMD-reconstituted 1,2-dimyristoyl-sn-glycero-3-phosphocholine/1,2-dimyristoyl-sn-glycero-3-phospho-(1'-rac-glycerol)/cholesterol lipid bilayers using electron spin resonance. We found that the FP-TMD increases the lipid order at all positions, which has a greater lipid ordering effect than the sum of the FP or TMD alone, and this effect reaches deeper into the membranes. Although HA-mediated membrane fusion is pH dependent, this combined effect is observed at both pH 5 and pH 7. In addition to increasing lipid order, multiple components are found for 5PC at increased concentration of FP-TMD, indicating that distinct domains are induced. However, the mutation of Gly1 in the FP and L187 in the TMD eliminates the perturbations, consistent with their fusogenic phenotypes. Electron spin resonance on spin-labeled peptides confirms these observations. We suggest that this interaction may provide a driving force in different stages of membrane fusion: initialization, transition from hemifusion stalk to transmembrane contact, and fusion pore formation.
|
|
|
Signal Transduction in Light–Oxygen–Voltage Receptors Lacking the Adduct-Forming Cysteine Residue
E.F. Yee, R.P. Diensthuber, A.T. Vaidya, P.P. Borbat, C. Engelhard, J.H. Freed, R. Bittl, A. Möglich, and B.R. Crane.
Nature Comm. 6 10079 (2015)
Supporting Information
<doi: 10.1038/ncomms10079>
PMID:
26648256
PMCID:
PMC4682037
|

|
|
ABSTRACT: Light–oxygen–voltage (LOV) receptors sense blue light through the photochemical generation of a covalent adduct between a flavin-nucleotide chromophore and a strictly conserved cysteine residue. Here we show that, after cysteine removal, the circadian-clock LOV-protein Vivid still undergoes light-induced dimerization and signalling because of flavin photoreduction to the neutral semiquinone (NSQ). Similarly, photoreduction of the engineered LOV histidine kinase YF1 to the NSQ modulates activity and downstream effects on gene expression. Signal transduction in both proteins hence hinges on flavin protonation, which is common to both the cysteinyl adduct and the NSQ. This general mechanism is also conserved by natural cysteine-less, LOV-like regulators that respond to chemical or photoreduction of their flavin cofactors. As LOV proteins can react to light even when devoid of the adduct-forming cysteine, modern LOV photoreceptors may have arisen from ancestral redox-active flavoproteins. The ability to tune LOV reactivity through photoreduction may have important implications for LOV mechanism and optogenetic applications.
|
|
|
Pulsed Dipolar Spectroscopy Reveals that Tyrosyl Radicals Are Generated in Both Monomers of the Cyclooxygenase-2 Dimer
B. J. Orlando, P. P. Borbat, E. R. Georgieva, J. H. Freed, and M. G. Malkowski.
Biochemistry 54 7309-7312 (2015)
Supporting Information
<doi: 10.1021/acs.biochem.5b00979>
PMID:
26636181
PMCID:
PMC4707933
|
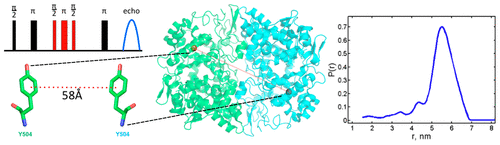
|
|
ABSTRACT: Cyclooxygenases (COXs) are heme-containing sequence homodimers that utilize tyrosyl radical-based catalysis to oxygenate substrates. Tyrosyl radicals are formed from a single turnover of substrate in the peroxidase active site generating an oxy-ferryl porphyrin cation radical intermediate that subsequently gives rise to a Tyr-385 radical in the cyclooxygenase active site and a Tyr-504 radical nearby. We have utilized double-quantum coherence (DQC) spectroscopy to determine the distance distributions between Tyr-385 and Tyr-504 radicals in COX-2. The distances obtained with DQC confirm that Tyr-385 and Tyr-504 radicals were generated in each monomer and accurately match the distances measured in COX-2 crystal structures.
|
|
|
Interaction of Spin-Labeled Lipid Membranes with Transition Metal Ions
B. Dzikovski, V. Livshits, and J.H. Freed
J. Phys. Chem. B 119, 13330-13346 (2015)
Supporting Information
<doi: 10.1021/acs.jpcb.5b08165>
PMID:
26490692
PMCID:
PMC4762260
|
|
|
ABSTRACT: The large values of spin relaxation enhancement (RE) for PC spin-labels in the phospholipid membrane induced by paramagnetic metal salts dissolved in the aqueous phase can be explained by Heisenberg spin exchange due to conformational fluctuations of the nitroxide group as a result of membrane fluidity, flexibility of lipid chains, and, possibly, amphiphilic nature of the nitroxide label. Whether the magnetic interaction occurs predominantly via Heisenberg spin exchange (Ni) or by the dipole–dipole (Gd) mechanism, it is essential for the paramagnetic ion to get into close proximity to the nitroxide moiety for efficient RE. For different salts of Ni the RE in phosphatidylcholine membranes follows the anionic Hofmeister series and reflects anion adsorption followed by anion-driven attraction of paramagnetic cations on the choline groups. This adsorption is higher for chaotropic ions, e.g., perchlorate. (A chaotropic agent is a molecule in water solution that can disrupt the hydrogen bonding network between water molecules.) However, there is no anionic dependence of RE for model membranes made from negatively charged lipids devoid of choline groups. We used Ni-induced RE to study the thermodynamics and electrostatics of ion/membrane interactions. We also studied the effect of membrane composition and the phase state on the RE values. In membranes with cholesterol a significant difference is observed between PC labels with nitroxide tethers long enough vs not long enough to reach deep into the membrane hydrophobic core behind the area of fused cholesterol rings. This study indicates one must be cautious in interpreting data obtained by PC labels in fluid membranes in terms of probing membrane properties at different immersion depths when it can be affected by paramagnetic species at the membrane surface.
|
|
|
Protein Dynamics in the Solid State from 2H NMR Line Shape Analysis. II. MOMD Applied to C–D and C–CD3 Probes
E. Meirovitch, Z. Liang, and J. H. Freed.
J. Phys. Chem. B 119 14022-14032 (2015)
<doi: 10.1021/acs.jpcb.5b07434>
PMID:
26402431
PMCID:
PMC4676681
|

|
|
ABSTRACT: Deuterium line shape analysis from mobile C–D and C–CD3 groups has emerged as a particularly useful tool for studying dynamics in the solid state. The theoretical models devised so far consist typically of sets of independent dynamic modes. Each such mode is simple and usually case-specific. In this scenario, model improvement entails adding yet another mode (thereby changing the overall model), comparison of different cases is difficult, and ambiguity is unavoidable. We recently developed the microscopic order macroscopic disorder (MOMD) approach as a single-mode alternative. In MOMD, the local spatial restrictions are expressed by an anisotropic potential, the local motion by a diffusion tensor, and the local molecular geometry by relative (magnetic and model-related) tensor orientations, all of adjustable symmetry. This approach provides a consistent method of analysis, thus resolving the issues above. In this study, we apply MOMD to PS-adsorbed LKα14 peptide and dimethylammonium tetraphenylborate (C–CD3 and N–CD3 dynamics, respectively), as well as HhaI methyltransferase target DNA and phase III of benzene-6-hexanoate (C–D dynamics). The success with fitting these four disparate cases, as well as the two cases in the previous report, demonstrates the generality of this MOMD-based approach. In this study, C–D and C–CD3 are both found to execute axial diffusion (rates R⊥ and R∥) in the presence of a rhombic potential given by the L = 2 spherical harmonics (coefficients c02 and c22). R⊥ (R∥) is in the 102–103 (104–105) s–1 range, and c02 and c22 are on the order of 2–3 kBT. Specific parameter values are determined for each mobile site. The diffusion and quadrupolar tensors are tilted at either 120° (consistent with trans–gauche isomerization) or nearly 110.5° (consistent with methyl exchange). Future prospects include extension of the MOMD formalism to include MAS, and application to 15N and 13C nuclei.
|
|
|
Mechanism of Influenza A M2 Transmembrane Domain Assembly in Lipid Membranes
E.R. Georgieva, P.P. Borbat, H.D. Norman, and J.H. Freed
Sci. Rep. 5, 11757 (2015).
Supporting Information
<doi: 10.1038/srep11757>
PMID:
26190831
PMCID:
PMC4507135
|
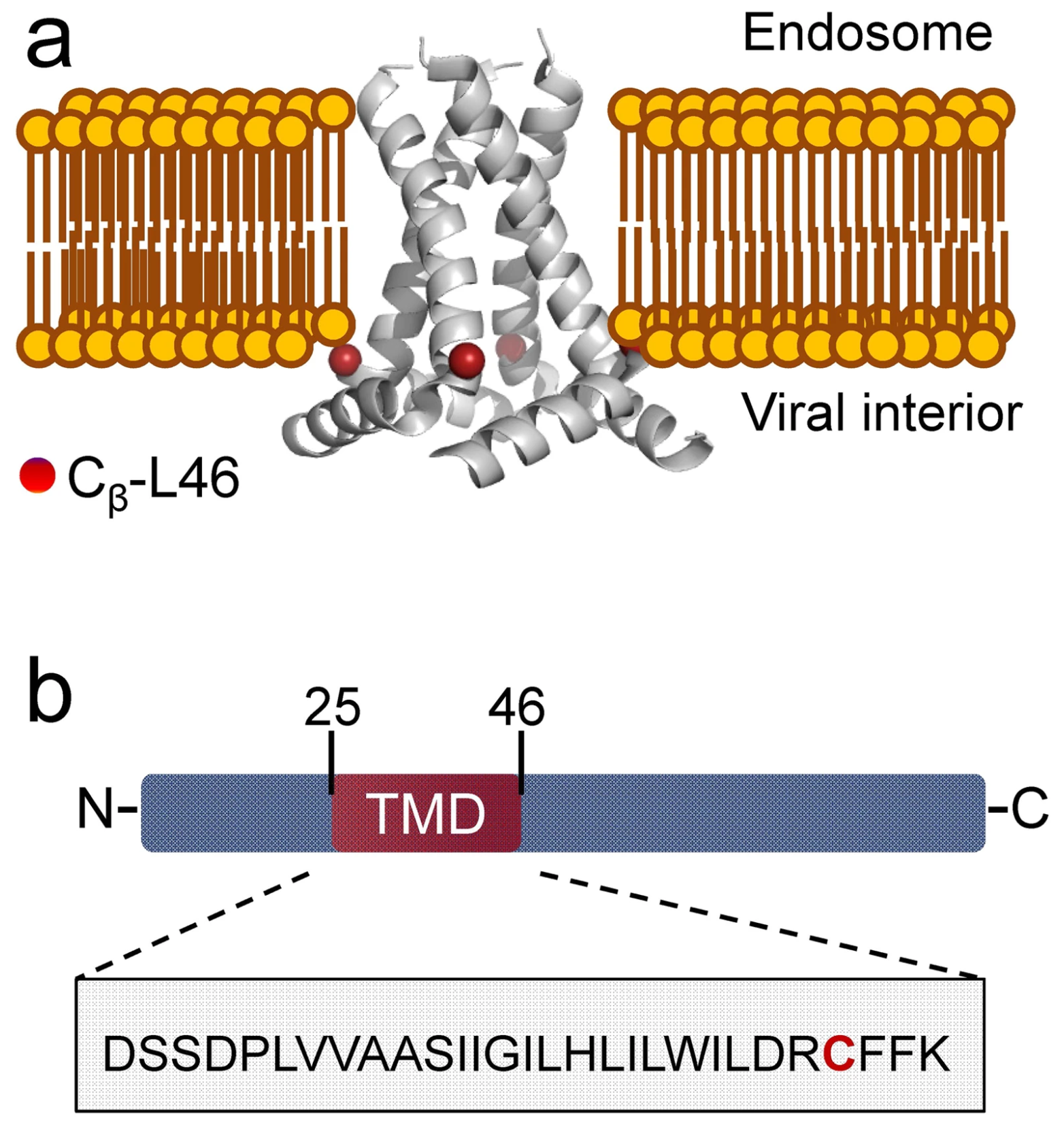
|
|
ABSTRACT: M2 from influenza A virus functions as an oligomeric proton channel essential for the viral cycle, hence it is a high-priority pharmacological target whose structure and functions require better understanding. We studied the mechanism of M2 transmembrane domain (M2TMD) assembly in lipid membranes by the powerful biophysical technique of double electron-electron resonance (DEER) spectroscopy. By varying the M2TMD-to-lipid molar ratio over a wide range from 1:18,800 to 1:160, we found that M2TMD exists as monomers, dimers and tetramers whose relative populations shift to tetramers with the increase of peptide-to-lipid (P/L) molar ratio. Our results strongly support the tandem mechanism of M2 assembly that is monomers-to-dimer then dimers-to-tetramer, since tight dimers are abundant at small P/L’s and thereafter they assemble as dimers of dimers in weaker tetramers. The stepwise mechanism found for a single-pass membrane protein oligomeric assembly should contribute to the knowledge of the association steps in membrane protein folding.
|
|
|
Preformed Soluble Chemoreceptor Trimers That Mimic Cellular Assembly States and Activate CheA Autophosphorylation
A.R. Greenswag, X. Li, P.P. Borbat, D. Samanta, K.J. Watts, J.H. Freed, and B.R. Crane
Biochemistry 54, 3454-3468 (2015).
Supporting Information
<doi: 10.1021/bi501570n>
PMID:
25967982
PMCID:
PMC4772074
|
|
|
ABSTRACT: Bacterial chemoreceptors associate with the histidine kinase CheA and coupling protein CheW to form extended membrane arrays that receive and transduce environmental signals. A receptor trimers-of-dimers resides at each vertex of the hexagonal protein lattice. CheA is fully activated and regulated when it is integrated into the receptor assembly. To mimic these states in solution, we have engineered chemoreceptor cytoplasmic kinase-control modules (KCMs) based on the Escherichia coli aspartate receptor Tar that are covalently fused and trimerized by a foldon domain (TarFO). Small-angle X-ray scattering, multi-angle light scattering, and pulsed-dipolar electron spin resonance spectroscopy of spin-labeled proteins indicate that the TarFO modules assemble into homogeneous trimers wherein the protein interaction regions closely associate at the end opposite to the foldon domains. The TarFO variants greatly increase the saturation levels of phosphorylated CheA (CheA-P), indicating that the association with a trimer of receptor dimers changes the fraction of active kinase. However, the rate constants for CheA-P formation with the Tar variants are low compared to those for autophosphorylation by free CheA, and net phosphotransfer from CheA to CheY does not increase commensurately with CheA autophosphorylation. Thus, the Tar variants facilitate slow conversion to an active form of CheA that then undergoes stable autophosphorylation and is capable of subsequent phosphotransfer to CheY. Free CheA is largely incapable of phosphorylation but contains a small active fraction. Addition of TarFO to CheA promotes a planar conformation of the regulatory domains consistent with array models for the assembly state of the ternary complex and different from that observed with a single inhibitory receptor. Introduction of TarFO into E. coli cells activates endogenous CheA to produce increased clockwise flagellar rotation, with the effects increasing in the presence of the chemotaxis methylation system (CheB/CheR). Overall, the TarFO modules demonstrate that trimerized signaling tips self-associate, bind CheA and CheW, and facilitate conversion of CheA to an active conformation.
|
|
|
Focus: Two-dimensional electron-electron double resonance and molecular motions: The challenge of higher frequencies
J.M. Franck, S. Chandrasekaran, B. Dzikovski, C.R. Dunnam, and J.H. Freed.
J. Chem. Phys. 142, 212302-212314 (2015).
<doi: 10.1063/1.4917322>
PMID:
26049420
PMCID:
PMC4443839
|
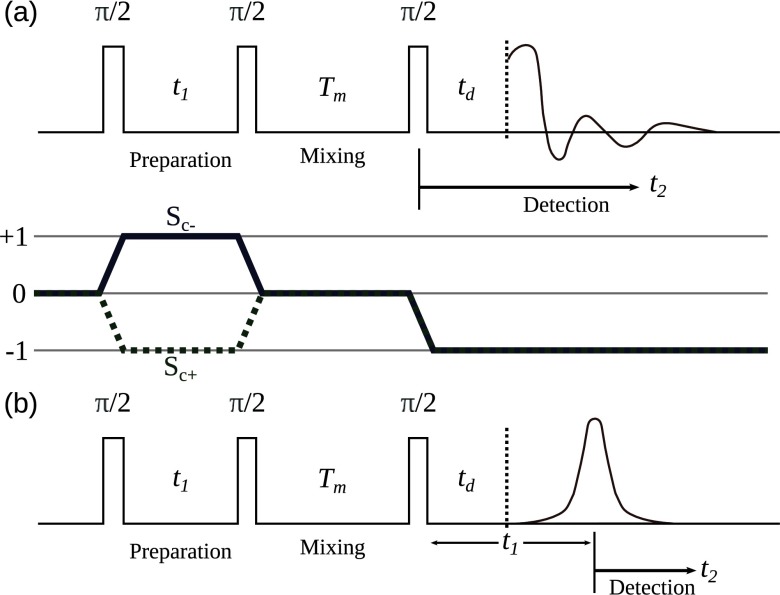
|
|
ABSTRACT: The development, applications, and current challenges of the pulsed ESR technique of two-dimensional Electron-Electron Double Resonance (2D ELDOR) are described. This is a three-pulse technique akin to 2D Exchange Nuclear Magnetic Resonance, but involving electron spins, usually in the form of spin-probes or spin-labels. As a result, it required the extension to much higher frequencies, i.e., microwaves, and much faster time scales, with π/2 pulses in the 2-3 ns range. It has proven very useful for studying molecular dynamics in complex fluids, and spectral results can be explained by fitting theoretical models (also described) that provide a detailed analysis of the molecular dynamics and structure. We discuss concepts that also appear in other forms of 2D spectroscopy but emphasize the unique advantages and difficulties that are intrinsic to ESR. Advantages include the ability to tune the resonance frequency, in order to probe different motional ranges, while challenges include the high ratio of the detection dead time vs. the relaxation times. We review several important 2D ELDOR studies of molecular dynamics. (1) The results from a spin probe dissolved in a liquid crystal are followed throughout the isotropic → nematic → liquid-like smectic → solid-like smectic → crystalline phases as the temperature is reduced and are interpreted in terms of the slowly relaxing local structure model. Here, the labeled molecule is undergoing overall motion in the macroscopically aligned sample, as well as responding to local site fluctuations. (2) Several examples involving model phospholipid membranes are provided, including the dynamic structural characterization of the boundary lipid that coats a transmembrane peptide dimer. Additionally, subtle differences can be elicited for the phospholipid membrane phases: liquid disordered, liquid ordered, and gel, and the subtle effects upon the membrane, of antigen cross-linking of receptors on the surface of plasma membrane, vesicles can be observed. These 2D ELDOR experiments are performed as a function of mixing time, Tm, i.e., the time between the second and third π/2 pulses, which provides a third dimension. In fact, a fourth dimension may be added by varying the ESR frequency/magnetic field combination. Therefore, (3) it is shown how continuous-wave multifrequency ESR studies enable the decomposition of complex dynamics of, e.g., proteins by virtue of their respective time scales. These studies motivate our current efforts that are directed to extend 2D ELDOR to higher frequencies, 95 GHz in particular (from 9 and 17 GHz), in order to enable multi-frequency 2D ELDOR. This required the development of quasi-optical methods for performing the mm-wave experiments, which are summarized. We demonstrate state-of-the-art 95 GHz 2D ELDOR spectroscopy through its ability to resolve the two signals from a spin probe dissolved in both the lipid phase and the coexisting aqueous phase. As current 95 GHz experiments are restricted by limited spectral coverage of the π/2 pulse, as well as the very short T2 relaxation times of the electron spins, we discuss how these limitations are being addressed.
|
|
|
Dynamic Nuclear Polarization of Membrane Proteins: Covalently Bound Spin-Labels at Protein-Protein Interfaces
B.J. Wylie, B.G. Dzikovski, S. Pawsey, M. Caporini, M. Rosay, J.H. Freed, A.E. McDermott.
J. Biomol. NMR 61, 361-367 (2015).
Supporting Information
<doi: 10.1007/s10858-015-9919-6>
PMID:
25828256
PMCID:
PMC4819240
|

|
|
ABSTRACT: We demonstrate that dynamic nuclear polarization of membrane proteins in lipid bilayers may be achieved using a novel polarizing agent: pairs of spin labels covalently bound to a protein of interest interacting at an intermolecular interaction surface. For gramicidin A, nitroxide tags attached to the N-terminal intermolecular interface region become proximal only when bimolecular channels forms in the membrane. We obtained signal enhancements of sixfold for the dimeric protein. The enhancement effect was comparable to that of a doubly tagged sample of gramicidin C, with intramolecular spin pairs. This approach could be a powerful and selective means for signal enhancement in membrane proteins, and for recognizing intermolecular interfaces.
|
|
|
Assembly States of FliM and FliG Within the Flagellar Switch Complex
R. Sircar, P.P. Borbat, M.J. Lynch, J. Bhatnagar, M.S. Beyersdorf, C.J. Halkides, J.H. Freed and B.R. Crane.
J. Mol. Biol. 427, 867-886 (2015).
Supporting Information
<doi: 10.1016/j.jmb.2014.12.009>
PMID:
25536293
PMCID:
PMC4323944
|
|
|
ABSTRACT: At the base of the bacterial flagella, a cytoplasmic rotor (the C-ring) generates torque and reverses rotation sense in response to stimuli. The bulk of the C-ring forms from many copies of the proteins FliG, FliM, and FliN, which together constitute the switch complex. To help resolve outstanding issues regarding C-ring architecture, we have investigated interactions between FliM and FliG from Thermotoga maritima with X-ray crystallography and pulsed dipolar ESR spectroscopy (PDS). A new crystal structure of an 11-unit FliG:FliM complex produces a large arc with a curvature consistent with the dimensions of the C-ring. Previously determined structures along with this new structure provided a basis to test switch complex assembly models. PDS combined with mutational studies and targeted cross-linking reveal that FliM and FliG interact through their middle domains to form both parallel and antiparallel arrangements in solution. Residue substitutions at predicted interfaces disrupt higher-order complexes that are primarily mediated by contacts between the C-terminal domain of FliG and the middle domain of a neighboring FliG molecule. Spin separations among multi-labeled components fit a self-consistent model that agree well with electron microscopy images of the C-ring. An activated form of the response regulator CheY destabilizes the parallel arrangement of FliM molecules to perturb FliG alignment in a process that may reflect the onset of rotation switching. These data suggest a model of C-ring assembly in which intermolecular contacts among FliG domains provide a template for FliM assembly and cooperative transitions.
|
|
|
Pulse Dipolar ESR of Doubly Labeled Mini TAR DNA and Its Annealing to Mini TAR RNA
Y. Sun, P.P. Borbat, V.M. Grigoryants, W.K. Myers, J.H. Freed, and C.P. Scholes.
Biophys. J. 108, 893-902 (2015).
Supporting Information
<doi: 10.1016/j.bpj.2014.12.028>
PMID:
25692594
PMCID:
PMC4336369
|

|
|
ABSTRACT: Pulse dipolar electron-spin resonance in the form of double electron electron resonance was applied to strategically placed, site-specifically attached pairs of nitroxide spin labels to monitor changes in the mini TAR DNA stem-loop structure brought on by the HIV-1 nucleocapsid protein NCp7. The biophysical structural evidence was at Ångstrom-level resolution under solution conditions not amenable to crystallography or NMR. In the absence of complementary TAR RNA, double labels located in both the upper and the lower stem of mini TAR DNA showed in the presence of NCp7 a broadened distance distribution between the points of attachment, and there was evidence for several conformers. Next, when equimolar amounts of mini TAR DNA and complementary mini TAR RNA were present, NCp7 enhanced the annealing of their stem-loop structures to form duplex DNA-RNA. When duplex TAR DNA-TAR RNA formed, double labels initially located 27.5 Å apart at the 3′- and 5′-termini of the 27-base mini TAR DNA relocated to opposite ends of a 27 bp RNA-DNA duplex with 76.5 Å between labels, a distance which was consistent with the distance between the two labels in a thermally annealed 27-bp TAR DNA-TAR RNA duplex. Different sets of double labels initially located 26–27 Å apart in the mini TAR DNA upper stem, appropriately altered their interlabel distance to ˜35 Å when a 27 bp TAR DNA-TAR RNA duplex formed, where the formation was caused either through NCp7-induced annealing or by thermal annealing. In summary, clear structural evidence was obtained for the fraying and destabilization brought on by NCp7 in its biochemical function as an annealing agent and for the detailed structural change from stem-loop to duplex RNA-DNA when complementary RNA was present.
|
|
|
Bacterial Chemoreceptor Dynamics Correlate With Activity State and Are Coupled Over Long Distances
D. Samanta, P.P. Borbat, B. Dzikovski, J.H. Freed, and B.R. Crane.
Proc. Natl. Acad. Sci. U. S. A. 112, 2455-2460 (2015).
Supporting Information
<doi: 10.1073/pnas.1414155112>
PMID:
25675479
PMCID:
PMC4345563
|

|
|
SIGNIFICANCE: Bacterial chemoreceptors are a key system for understanding how conformational signals propagate over large distances in transmembrane signaling. We have applied pulsed dipolar ESR spectroscopy of spin-labeled receptors to correlate conformation and dynamics with activity state. We find that the receptor cytoplasmic domain behaves as one large dynamically coupled system, in which activation signals destabilize membrane proximal regions but stabilize the most distal protein interaction tip. Inhibitory signals or adaptations of the receptor through chemical modification produce the opposite changes in conformational properties. This reciprocal coupling of conformational stability provides a versatile mechanism for sending signals throughout large modular proteins.
ABSTRACT: Dynamics are hypothesized to play an important role in the transmission of signals across membranes by receptors. Bacterial chemoreceptors are long helical proteins that consist of a periplasmic ligand-binding domain; a transmembrane region; a cytoplasmic HAMP (histidine kinase, adenylyl cyclases, methyl-accepting chemotaxis proteins, and phosphatases) domain; and a kinase-control module (KCM). The KCM is further composed of adaptation, hinge, and protein interaction regions (PIRs), the latter of which binds the histidine kinase CheA and adaptor CheW. Fusions of the Escherichia coli aspartate receptor KCM to HAMP domains of defined structure (H1-Tar vs. H1-2-Tar) give opposite responses in phosphotransfer and cellular assays, despite similar binding to CheA and CheW. Pulsed dipolar ESR spectroscopy (PDS) of these isolated on and off dimeric effectors reveals that, in the kinase-on state, the HAMP is more conformationally destabilized compared with the PIR, whereas in the kinase-off state, the HAMP is more compact, and the PIR samples a greater breadth of conformations. On and off HAMP states produce different conformational effects at the KCM junction, but these differences decrease through the adaptation region and into the hinge only to return with the inverted relationship in the PIR. Continuous wave–ESR of the spin-labeled proteins confirms that broader PDS distance distributions correlate with increased rates of dynamics. Conformational breadth in the adaptation region changes with charge alterations caused by modification enzymes. Activating modifications broaden the HAMP conformational ensemble but correspondingly, compact the PIR. Thus, chemoreceptors behave as coupled units, in which dynamics in regions proximal and distal to the membrane change coherently but with opposite sign.
|
|
|
Transport Domain Unlocking Sets the Uptake Rate of an Aspartate Transporter
N. Akyuz, E.R. Georgieva, Z. Zhou, S. Stolzenberg, M.A. Cuendet, G. Khelashvili, R.B. Altman, D.S. Terry, J.H. Freed, H. Weinstein, O. Boudker, and S.C. Blanchard.
Nature 518, 68-73 (2015).
|

|
|
Transport Domain Unlocking Sets the Uptake Rate of an Aspartate Transporter
N. Akyuz, E.R. Georgieva, Z. Zhou, S. Stolzenberg, M.A. Cuendet, G. Khelashvili, R.B. Altman, D.S. Terry, J.H. Freed, H. Weinstein, O. Boudker, and S.C. Blanchard.
Nature 518, 68-73 (2015).
Supporting Information
<doi: 10.1038/nature14158>
PMID:
25652997
PMCID:
PMC4351760
|
|
|
ABSTRACT: Glutamate transporters terminate neurotransmission by clearing synaptically released glutamate from the extracellular space, allowing repeated rounds of signalling and preventing glutamate-mediated excitotoxicity. Crystallographic studies of a glutamate transporter homologue from the archaeon Pyrococcus horikoshii, GltPh, showed that distinct transport domains translocate substrates into the cytoplasm by moving across the membrane within a central trimerization scaffold. Here we report direct observations of these 'elevator-like' transport domain motions in the context of reconstituted proteoliposomes and physiological ion gradients using single-molecule fluorescence resonance energy transfer (smFRET) imaging. We show that GltPh bearing two mutations introduced to impart characteristics of the human transporter exhibits markedly increased transport domain dynamics, which parallels an increased rate of substrate transport, thereby establishing a direct temporal relationship between transport domain motion and substrate uptake. Crystallographic and computational investigations corroborated these findings by revealing that the 'humanizing' mutations favour structurally 'unlocked' intermediate states in the transport cycle exhibiting increased solvent occupancy at the interface between the transport domain and the trimeric scaffold.
|
|
|
Protein Dynamics in the Solid State from 2H NMR Line Shape Analysis: A Consistent Perspective
E. Meirovitch, Z. Liang, and J.H. Freed.
J. Phys. Chem B 119, 2857-2868 (2015).
<doi: 10.1021/jp511386b>
PMID:
25594631
PMCID:
PMC4358757
|

|
|
ABSTRACT: Deuterium line shape analysis of CD3 groups has emerged as a particularly useful tool for studying microsecond–millisecond protein motions in the solid state. The models devised so far consist of several independently conceived simple jump-type motions. They are comprised of physical quantities encoded in their simplest form; improvements are only possible by adding yet another simple motion, thereby changing the model. The various treatments developed are case-specific; hence comparison among the different systems is not possible. Here we develop a new methodology for 2H NMR line shape analysis free of these limitations. It is based on the microscopic-order-macroscopic-disorder (MOMD) approach. In MOMD motions are described by diffusion tensors, spatial restrictions by potentials/ordering tensors, and geometric features by relative tensor orientations. Jump-type motions are recovered in the limit of large orientational potentials. Model improvement is accomplished by monitoring the magnitude, symmetry, and orientation of the various tensors. The generality of MOMD makes possible comparison among different scenarios. CD3 line shapes from the Chicken Villin Headpiece Subdomain and the Streptomyces Subtilisin Inhibitor are used as experimental examples. All of these spectra are reproduced by using rhombic local potentials constrained for simplicity to be given by the L = 2 spherical harmonics, and by axial diffusion tensors. Potential strength and rhombicity are found to be ca. 2–3 kBT. The diffusion tensor is tilted at 120° from the C–CD3 axis. The perpendicular (parallel) correlation times for local motion are 0.1–1.0 ms (3.3–30 μs). Activation energies in the 1.1–8.0 kcal/mol range are estimated. Future prospects include extension to the 2H relaxation limit, application to the 15N and 13C NMR nuclei, and accounting for collective motions and anisotropic media.
|
|
|
Aggregation Propensities of Superoxide Dismutase G93 Hotspot Mutants Mirror ALS Clinical Phenotypes
A.J. Pratt, D.S. Shin, G.E. Merz, R.P. Rambo, W.A. Lancaster, K.N. Dyer, P.P. Borbat, F.L. Poole II, M.W.W. Adams, J.H. Freed, B.R. Crane, J.A. Tainer, and E.D. Getzoff.
Proc. Natl. Acad. Sci. U. S. A. 111, E4568-E4576 (2014).
|
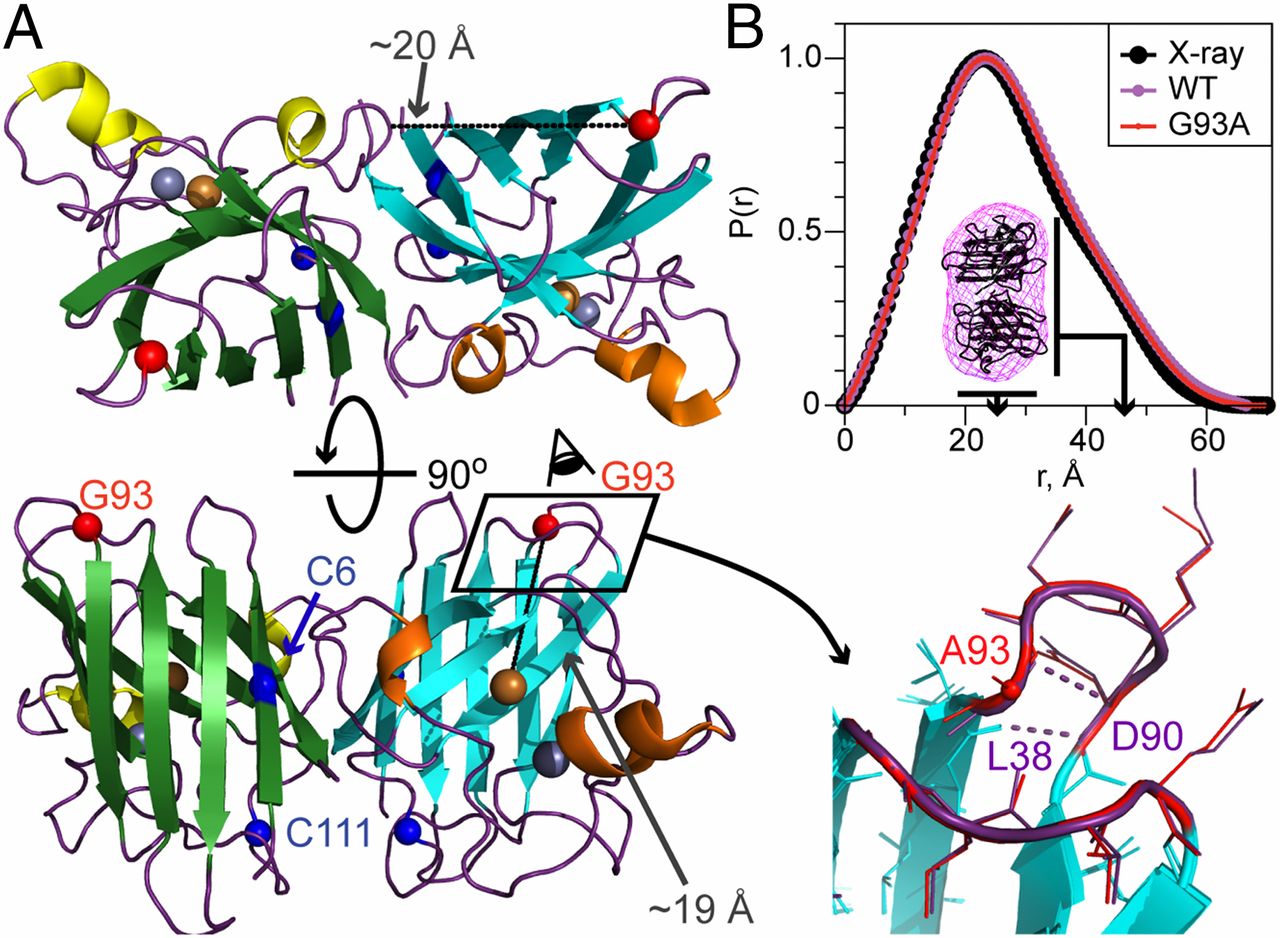
|
|
Aggregation Propensities of Superoxide Dismutase G93 Hotspot Mutants Mirror ALS Clinical Phenotypes
A.J. Pratt, D.S. Shin, G.E. Merz, R.P. Rambo, W.A. Lancaster, K.N. Dyer, P.P. Borbat, F.L. Poole II, M.W.W. Adams, J.H. Freed, B.R. Crane, J.A. Tainer, and E.D. Getzoff.
Proc. Natl. Acad. Sci. U. S. A. 111, E4568-E4576 (2014).
Supporting Information
<doi: 10.1073/pnas.1308531111>
PMID:
25316790
PMCID:
PMC4217430
|
|
|
SIGNIFICANCE: Mutations in human Cu, Zn superoxide dismutase (SOD) cause the motor neuron disease ALS. To better understand why, we compared the aggregation, metal binding, and conformational dynamics of normal and mutant SOD proteins by using the biophysical techniques of X-ray scattering, inductively coupled plasma MS, and ESR spectroscopy. For SOD proteins with defects at a mutational hotspot, we found that copper deficiency, flexibility, and aggregation paralleled clinical severity in ALS patients. These data support a unifying protein framework destabilization mechanism for SOD-linked ALS and thereby point to potential therapies for this lethal condition with few treatment options.
ABSTRACT: Protein framework alterations in heritable Cu, Zn superoxide dismutase (SOD) mutants cause misassembly and aggregation in cells affected by the motor neuron disease ALS. However, the mechanistic relationship between superoxide dismutase 1 (SOD1) mutations and human disease is controversial, with many hypotheses postulated for the propensity of specific SOD mutants to cause ALS. Here, we experimentally identify distinguishing attributes of ALS mutant SOD proteins that correlate with clinical severity by applying solution biophysical techniques to six ALS mutants at human SOD hotspot glycine 93. A small-angle X-ray scattering (SAXS) assay and other structural methods assessed aggregation propensity by defining the size and shape of fibrillar SOD aggregates after mild biochemical perturbations. Inductively coupled plasma MS quantified metal ion binding stoichiometry, and pulsed dipolar ESR spectroscopy evaluated the Cu2+ binding site and defined cross-dimer copper–copper distance distributions. Importantly, we find that copper deficiency in these mutants promotes aggregation in a manner strikingly consistent with their clinical severities. G93 mutants seem to properly incorporate metal ions under physiological conditions when assisted by the copper chaperone but release copper under destabilizing conditions more readily than the WT enzyme. Altered intradimer flexibility in ALS mutants may cause differential metal retention and promote distinct aggregation trends observed for mutant proteins in vitro and in ALS patients. Combined biophysical and structural results test and link copper retention to the framework destabilization hypothesis as a unifying general mechanism for both SOD aggregation and ALS disease progression, with implications for disease severity and therapeutic intervention strategies.
|
|
|
Copper-Based Pulsed Dipolar ESR Spectroscopy as a Probe of Protein Conformation Linked to Disease States
G.E. Merz, P.P. Borbat, A.J. Pratt, E.D. Getzoff, J.H. Freed, and B.R. Crane.
Biophys. J. 107, 1669-1674 (2014).
Supporting Information
<doi: 10.1016/j.bpj.2014.07.068>
PMID:
25296320
PMCID:
PMC4190658
|

|
|
ABSTRACT: We demonstrate the ability of pulsed dipolar electron spin resonance (ESR) spectroscopy (PDS) to report on the conformation of Cu-Zn superoxide dismutase (SOD1) through the sensitive measurement of dipolar interactions between inherent Cu2+ ions. Although the extent and the anisotropy of the Cu ESR spectrum provides challenges for PDS, Ku-band (17.3 GHz) double electron-electron resonance and double-quantum coherence variants of PDS coupled with distance reconstruction methods recover Cu-Cu distances in good agreement with crystal structures. Moreover, Cu-PDS measurements expose distinct differences between the conformational properties of wild-type SOD1 and a single-residue variant (I149T) that leads to the disease amyotrophic lateral sclerosis (ALS). The I149T protein displays a broader Cu-Cu distance distribution within the SOD1 dimer compared to wild-type. In a nitroxide (NO)-labeled sample, distance distributions obtained from Cu-Cu, Cu-NO, and NO-NO separations reveal increased structural heterogeneity within the protein and a tendency for mutant dimers to associate. In contrast, perturbations caused by the ALS mutation are completely masked in the crystal structure of I149T. Thus, PDS readily detects alterations in metalloenzyme solution properties not easily deciphered by other methods and in doing so supports the notion that increased range of motion and associations of SOD1 ALS variants contribute to disease progression.
|
|
|
Tau Binds to Lipid Membrane Surfaces via Short Amphipathic Helices Located in Its Microtubule-Binding Repeats
E.R. Georgieva, S. Xiao, P.P. Borbat, J.H. Freed, and D. Eliezer.
Biophys. J. 107, 1441-1452 (2014).
Supporting Information
<doi: 10.1016/j.bpj.2014.07.046>
PMID:
25229151
PMCID:
PMC4167292
|
|
|
ABSTRACT: Tau is a microtubule-associated protein that is genetically linked to dementia and linked to Alzheimer's disease via its presence in intraneuronal neurofibrillary tangle deposits, where it takes the form of aggregated paired helical and straight filaments. Although the precise mechanisms by which tau contributes to neurodegeneration remain unclear, tau aggregation is commonly considered to be a critical component of tau-mediated pathogenicity. Nevertheless, the context in which tau aggregation begins in vivo is unknown. Tau is enriched in membrane-rich neuronal structures such as axons and growth cones, and can interact with membranes both via intermediary proteins and directly via its microtubule-binding domain (MBD). Membranes efficiently facilitate tau aggregation in vitro, and may therefore provide a physiologically relevant context for nucleating tau aggregation in vivo. Furthermore, tau-membrane interactions may potentially play a role in tau's poorly understood normal physiological functions. Despite the potential importance of direct tau-membrane interactions for tau pathology and physiology, the structural mechanisms that underlie such interactions remain to be elucidated. Here, we employ electron spin resonance spectroscopy to investigate the secondary and long-range structural properties of the MBD of three-repeat tau isoforms when bound to lipid vesicles and membrane mimetics. We show that the membrane interactions of the tau MBD are mediated by short amphipathic helices formed within each of the MBD repeats in the membrane-bound state. To our knowledge, this is the first detailed elucidation of helical tau structure in the context of intact lipid bilayers. We further show, for the first time (to our knowledge), that these individual helical regions behave as independent membrane-binding sites linked by flexible connecting regions. These results represent the first (to our knowledge) detailed structural view of membrane-bound tau and provide insights into potential mechanisms for membrane-mediated tau aggregation. Furthermore, the results may have implications for the structural basis of tau-microtubule interactions and microtubule-mediated tau aggregation.
|
|
|
An Iron-Sulfur Cluster in the Polymerase Domain of Yeast DNA Polymerase ε
R. Jain, E.S. Vanamee, B.G. Dzikovski, A. Buku, R.E. Johnson, L. Prakash, S. Prakash, A.K. Aggarwal.
J. Mol. Biol. 426 301-308 (2014)
Supporting Information
<doi: 10.1016/j.jmb.2013.10.015>
PMID:
24144619
PMCID:
PMC4061903
|

|
|
ABSTRACT: DNA polymerase ε (Polε) is a multi-subunit polymerase that contributes to genomic stability via its roles in leading strand replication and the repair of damaged DNA. Polε from Saccharomyces cerevisiae is composed of four subunits—Pol2, Dpb2, Dpb3, and Dpb4. Here, we report the presence of a [Fe-S] cluster directly within the active polymerase domain of Pol2 (residues 1–1187). We show that binding of the [Fe-S] cluster is mediated by cysteines in an insertion (Pol2ins) that is conserved in Pol2 orthologs but is absent in the polymerase domains of Polα, Polδ, and Polζ. We also show that the [Fe-S] cluster is required for Pol2 polymerase activity but not for its exonuclease activity. Collectively, our work suggests that Polε is perhaps more sensitive than other DNA polymerases to changes in oxidative stress in eukaryotic cells.
|
|
|
Dph3 Is an Electron Donor for Dph1-Dph2 in the First Step of Eukaryotic Diphthamide Biosynthesis
M. Dong, X. Su, B. Dzikovski, E.E. Dando, X. Zhu, J. Du, J.H. Freed, and H. Lin.
J. Am. Chem. Soc. 136, 1754-1757 (2014).
Supporting Information
<doi: 10.1021/ja4118957>
PMID:
24422557
PMCID:
PMC3985478
|

|
|
ABSTRACT: Diphthamide, the target of diphtheria toxin, is a unique posttranslational modification on translation elongation factor 2 (EF2) in archaea and eukaryotes. The biosynthesis of diphthamide was proposed to involve three steps. The first step is the transfer of the 3-amino-3-carboxypropyl group from S-adenosyl-L-methionine (SAM) to the histidine residue of EF2, forming a C–C bond. Previous genetic studies showed this step requires four proteins in eukaryotes, Dph1–Dph4. However, the exact molecular functions for the four proteins are unknown. Previous study showed that Pyrococcus horikoshii Dph2 (PhDph2), a novel iron-sulfur cluster-containing enzyme, forms a homodimer and is sufficient for the first step of diphthamide biosynthesis in vitro. Here we demonstrate by in vitro reconstitution that yeast Dph1 and Dph2 form a complex (Dph1-Dph2) that is equivalent to the homodimer of PhDph2 and is sufficient to catalyze the first step in vitro in the presence of dithionite as the reductant. We further demonstrate that yeast Dph3 (also known as KTI11), a CSL-type zinc finger protein, can bind iron and in the reduced state can serve as an electron donor to reduce the Fe-S cluster in Dph1-Dph2. Our study thus firmly establishes the functions for three of the proteins involved in eukaryotic diphthamide biosynthesis. For most radical SAM enzymes in bacteria, flavodoxins and flavodoxin reductases are believed to serve as electron donors for the Fe-S clusters. The finding that Dph3 is an electron donor for the Fe-S clusters in Dph1-Dph2 is thus interesting and opens up new avenues of research on electron transfer to Fe-S proteins in eukaryotic cells.
|
|
|
HIV gp41 Fusion Peptide Increases Membrane Ordering in a Cholesterol-Dependent Fashion
A.L. Lai and J.H. Freed.
Biophys. J. 106, 172-181 (2014).
Supporting Information
<doi: 10.1016/j.bpj.2013.11.027>
PMID:
24411249
PMCID:
PMC3907210
|
|
|
ABSTRACT: Fusion between viral envelopes and host cell membranes, which is mediated by special glycoproteins anchored on the viral membrane, is required for HIV viral entry and infection. The HIV gp41 fusion peptide (FP), which initiates membrane fusion, adopts either an α-helical or β-sheeted structure depending on the cholesterol concentration. We used phosphocholine spin labels on the lipid headgroup and different positions on the acyl chain to detect its perturbation on lipid bilayers containing different cholesterol concentrations by electron-spin resonance. Our findings were as follows. 1), gp41 FP affects the lipid order in the same manner as previously shown for influenza hemagglutinin FP, i.e., it has a cooperative effect versus the peptide/lipid ratio, supporting our hypothesis that membrane ordering is a common prerequisite for viral membrane fusion. 2), gp41 FP induces membrane ordering in all lipid compositions studied, whereas a nonfusion mutant FP perturbs lipid order to a significantly smaller extent. 3), In high-cholesterol-containing lipid bilayers, where gp41 FP is in the β-aggregation conformation, its effect on the lipid ordering reaches deeper into the bilayer. The different extent to which the two conformers perturb is correlated with their fusogenicity. The possible role of the two conformers in membrane fusion is discussed.
|
|
|
Architecture of the Soluble Receptor Aer2 Indicates an In-Line Mechanism for PAS and HAMP Domain Signaling
M.V. Airola, D. Huh, N. Sukomon, J. Widom, R. Sircar, P.P. Borbat, J.H. Freed, K.J. Watts, and B.R. Crane.
J. Mol. Biol. 425, 886-901 (2013).
Supporting Information
<doi: 10.1016/j.jmb.2012.12.011>
PMID:
23274111
PMCID:
PMC3577987
|
|
|
ABSTRACT: Bacterial receptors typically contain modular architectures with distinct functional domains that combine to send signals in response to stimuli. Although the properties of individual components have been investigated in many contexts, there is little information about how diverse sets of modules work together in full-length receptors. Here, we investigate the architecture of Aer2, a soluble gas-sensing receptor that has emerged as a model for PAS (Per–Arnt–Sim) and poly-HAMP (histidine kinase–adenylyl cyclase–methyl-accepting chemotaxis protein–phosphatase) domain signaling. The crystal structure of the heme-binding PAS domain in the ferric, ligand-free form, in comparison to the previously determined cyanide-bound state, identifies conformational changes induced by ligand binding that are likely essential for the signaling mechanism. Heme-pocket alternations share some similarities with the heme-based PAS sensors FixL and EcDOS but propagate to the Iβ strand in a manner predicted to alter PAS–PAS associations and the downstream HAMP junction within full-length Aer2. Small-angle X-ray scattering of PAS and poly-HAMP domain fragments of increasing complexity allow unambiguous domain assignments and reveal a linear quaternary structure. The Aer2 PAS dimeric crystal structure fits well within ab initio small-angle X-ray scattering molecular envelopes, and pulsed dipolar ESR measurements of inter-PAS distances confirm the crystallographic PAS arrangement within Aer2. Spectroscopic and pull-down assays fail to detect direct interactions between the PAS and HAMP domains. Overall, the Aer2 signaling mechanism differs from the Escherichia coli Aer paradigm, where side-on PAS–HAMP contacts are key. We propose an in-line model for Aer2 signaling, where ligand binding induces alterations in PAS domain structure and subunit association that is relayed through the poly-HAMP junction to downstream domains.
|
|
|
HAMP Domain Conformers that Propagate Opposite Signals in Bacterial Chemoreceptors
M.V. Airola, N. Sukomon, D. Samanta, P.P. Borbat, J.H. Freed, K.J. Watts, and B.R. Crane.
PLoS Biol. 11, e1001479, (2013).
Supporting Information
<doi: 10.1371/journal.pbio.1001479>
PMID:
23424282
PMCID:
PMC3570549
|
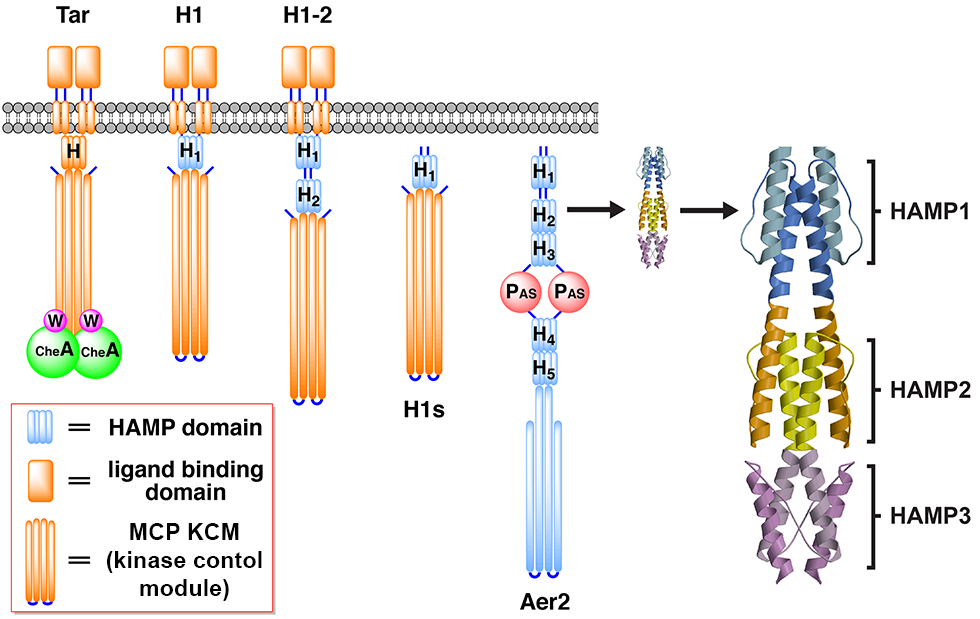
|
|
ABSTRACT: HAMP domains are signal relay modules in >26,000 receptors of bacteria, eukaryotes, and archaea that mediate processes involved in chemotaxis, pathogenesis, and biofilm formation. We identify two HAMP conformations distinguished by a four- to two-helix packing transition at the C-termini that send opposing signals in bacterial chemoreceptors. Crystal structures of signal-locked mutants establish the observed structure-to-function relationships. Pulsed dipolar electron spin resonance spectroscopy of spin-labeled soluble receptors active in cells verify that the crystallographically defined HAMP conformers are maintained in the receptors and influence the structure and activity of downstream domains accordingly. Mutation of HR2, a key residue for setting the HAMP conformation and generating an inhibitory signal, shifts HAMP structure and receptor output to an activating state. Another HR2 variant displays an inverted response with respect to ligand and demonstrates the fine energetic balance between "on" and "off" conformers. A DExG motif found in membrane proximal HAMP domains is shown to be critical for responses to extracellular ligand. Our findings directly correlate in vivo signaling with HAMP structure, stability, and dynamics to establish a comprehensive model for HAMP-mediated signal relay that consolidates existing views on how conformational signals propagate in receptors. Moreover, we have developed a rational means to manipulate HAMP structure and function that may prove useful in the engineering of bacterial taxis responses.
|
|
|
Conformational Ensemble of the Sodium-Coupled Aspartate Transporter
E.R. Georgieva, P.P. Borbat, C. Ginter, J.H. Freed, and O. Boudker.
Nat. Struct. Mol. Biol. 20, 215-221 (2013).
Supporting Information
<doi: 10.1038/nsmb.2494>
PMID:
23334289
PMCID:
PMC3565060
|

|
|
ABSTRACT: Sodium and aspartate symporter from Pyrococcus horikoshii, GltPh, is a homolog of the mammalian glutamate transporters, homotrimeric integral membrane proteins that control neurotransmitter levels in brain synapses. These transporters function by alternating between outward-facing and inward-facing states, in which the substrate binding site is oriented toward the extracellular space and the cytoplasm, respectively. Here we used double electron-electron resonance (DEER) spectroscopy to probe the structure and the state distribution of the subunits in the trimer in distinct hydrophobic environments of detergent micelles and lipid bilayers. Our experiments reveal a conformational ensemble of protomers that sample the outward-facing and inward-facing states with nearly equal probabilities, indicative of comparable energies, and independently of each other. On average, the distributions varied only modestly in detergent and in bilayers, but in several mutants unique conformations were stabilized by the latter.
|
|
|
Pulse Dipolar Electron Spin Resonance: Distance Measurements
P.P. Borbat and J.H. Freed.
In Structural Information from Spin-Labels and Intrinsic Paramagnetic Centers in the Biosciences. Structure and Bonding. Vol. 152. J. Harmer and C. Timmel, Eds.
Springer: Heidelberg, Germany; New York, USA, 2013; pp. 1-82.
|

|
|
Pulse Dipolar Electron Spin Resonance: Distance Measurements
P.P. Borbat and J.H. Freed.
In Structural Information from Spin-Labels and Intrinsic Paramagnetic Centers in the Biosciences. Structure and Bonding. Vol. 152. J. Harmer and C. Timmel, Eds.
Springer: Heidelberg, Germany; New York, USA, 2013; pp. 1-82.
<doi: 10.1007/430_2012_82>
PMID: [None - Book] PMCID:
[None - Book]
|
|
|
ABSTRACT: In recent years electron spin resonance (ESR) has provided the means to obtain structural constraints in the field of structural biology on the nanoscale by measuring distances between paramagnetic species, which usually have been nitroxide spin-labels. These ESR methods enable the measurement of distances over the wide range from ca. 6–10 Å to nearly 90 Å. While cw methods may be used for the shortest distances, it is the pulse methods that enable this wide range, as well as determination of the distributions in distance. In this chapter we first describe the underlying theoretical concepts for understanding the principal pulse methods of double quantum coherence (DQC)-ESR and double-electron–electron-resonance (DEER), which we collectively refer to as Pulse-Dipolar ESR Spectroscopies (PDS). We then provide technical aspects of pulse ESR spectrometers required for high quality PDS studies. This is followed by an extensive description of sensitivity considerations in PDS, based largely upon our highly sensitive 17.3 GHz pulse spectrometer at ACERT. This description also includes a comparison of the effectiveness of the respective PDS pulse methods. In addition, the newer methods of 5-pulse DEER, which enables longer distances to be measured than by standard DEER, and 2D-DQC, which provides a convenient mapping for studying orientational coherence between spin labels and their interspin vector, are described.
|
|
|
Improved Sensitivity for Long-Distance Measurements in Biomolecules: Five-Pulse Double Electron-Electron Resonance
P.P. Borbat, E.R. Georgieva, and J.H. Freed.
J. Phys. Chem. Lett. 4, 170-175 (2013).
Supporting Information
<doi: 10.1021/jz301788n>
PMID:
23301118
PMCID:
PMC3538160
|

|
|
ABSTRACT: We describe significantly improved long-distance measurements in biomolecules by use of the new multipulse double electron–electron spin resonance (DEER) illustrated with the example of a five-pulse DEER sequence. In this sequence, an extra pulse at the pump frequency is used compared with standard four-pulse DEER. The position of the extra pulse is fixed relative to the three pulses of the detection sequence. This significantly reduces the effect of nuclear spin-diffusion on the electron-spin phase relaxation, thereby enabling longer dipolar evolution times that are required to measure longer distances. Using spin-labeled T4 lysozyme at a concentration less than 50 μM, as an example, we show that the evolution time increases by a factor of 1.8 in protonated solution and 1.4 in deuterated solution to 8 and 12 μs, respectively, with the potential to increase them further. This enables a significant increase in the measurable distances, improved distance resolution, or both.
|
|
|
Locating a Lipid at the Portal to the Lipoxygenase Active Site
B.J. Gaffney, M. Bradshaw, S. Frausto, F. Wu, J.H. Freed, and P.P. Borbat.
Biophysical J. 103, 2134-2144, (2012).
Supporting Information
<doi: 10.1016/j.bpj.2012.10.002>
PMID:
23200047
PMCID:
PMC3512035
|
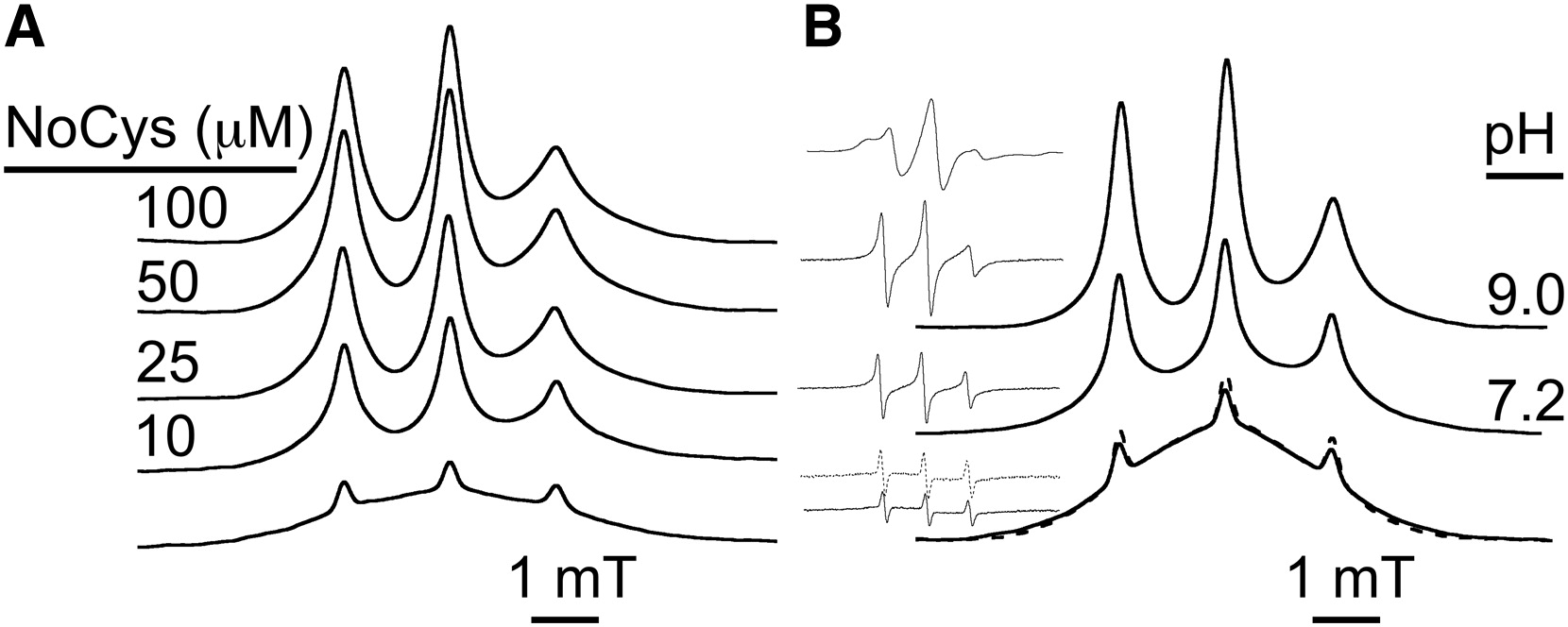
|
|
ABSTRACT: Lipoxygenase enzymes initiate diverse signaling pathways by specifically directing oxygen to different carbons of arachidonate and other polyunsaturated acyl chains, but structural origins of this specificity have remained unclear. We therefore determined the nature of the lipoxygenase interaction with the polar-end of a paramagnetic lipid by electron paramagnetic resonance spectroscopy. Distances between selected grid points on soybean seed lipoxygenase-1 (SBL1) and a lysolecithin spin-labeled on choline were measured by pulsed (electron) dipolar spectroscopy. The protein grid was designed by structure-based modeling so that five natural side chains were replaced with spin labels. Pairwise distances in 10 doubly spin-labeled mutants were examined by pulsed dipolar spectroscopy, and a fit to the model was optimized. Finally, experimental distances between the lysolecithin spin and each single spin site on SBL1 were also obtained. With these 15 distances, distance geometry localized the polar-end and the spin of the lysolecithin to the region between the two domains in the SBL1 structure, nearest to E236, K260, Q264, and Q544. Mutation of a nearby residue, E256A, relieved the high pH requirement for enzyme activity of SBL1 and allowed lipid binding at pH 7.2. This general approach could be used to locate other flexible molecules in macromolecular complexes.
|
|
|
Membrane Fluidity
B. Dzikovski and J.H. Freed.
In Encyc. Biophys.; G.C.K. Roberts, Ed.
Springer Verlag: Berlin, Germany, 2013; pp 1440-1446.
|

|
|
Membrane Fluidity
B. Dzikovski and J.H. Freed.
In Encyc. Biophys.; G.C.K. Roberts, Ed.
Springer Verlag: Berlin, Germany, 2013; pp 1440-1446.
<doi: 10.1007/978-3-642-16712-6_546>
PMID: [None - Book] PMCID:
[None - Book]
|
|
|
ABSTRACT: In 1972, Singer and Nicolson (Singer and Nicolson 1972) suggested the so-called fluid mosaic model of the biological membrane (Fig. 1). This useful hypothesis explained many phenomena occurring in model and biological membranes. According to this model, membrane proteins and other membrane-embedded compounds are suspended in a two-dimensional fluid formed by phospholipids. This fluid state of membrane lipids is critical for membrane function. It allows, for example, free diffusion and equal distribution of new cell-synthesized lipids and proteins; lateral diffusion of proteins and other molecules in signaling events and other membrane reactions; membrane fusion, that is, fusion of vesicles with organelles; separation of membranes during cell division; etc.
Membrane fluidity, which describes the ease of movement for molecules in the membrane environment, is a general concept that lacks a precise definition. It is much broader than the strict physical definition of fluidity as the reciprocal of viscosity in the case of isotropic liquids. In general, "membrane fluidity" implies various anisotropic motions, which contribute to the mobility of components of a membrane.
The lipid membrane, as a whole, shows a unique combination of fluidity and rigidity. In terms of the solubility and diffusion of small nonpolar molecules, the membrane behaves very much like an oil drop. In contrast, the translational diffusion constants of lipids and proteins in membranes are characteristic of media with the viscosity over two orders of magnitude greater than that of oil such as hexadecane. Also, in most cases, the membrane represents an impermeable barrier for ions and other hydrophilic compounds.
Figure 2 shows characteristic frequencies (reciprocal of characteristic times) of different kinds of molecular motions in the membrane in comparison to frequency ranges in which various spectroscopic techniques are sensitive to molecular motion (Gennis 1989).
|
|
|
The Internal Dynamics of Mini c TAR DNA Probed by Electron Paramagnetic Resonance of Nitroxide Spin-Labels at the Lower Stem, the Loop, and the Bulge
Y. Sun, Z. Zhang, V.M. Grigoryants, W.K. Myers, F. Liu, and K. Earle, J.H. Freed, C.P. Scholes
Biochemistry 51, 8530-8541 (2012).
|

|
|
The Internal Dynamics of Mini c TAR DNA Probed by Electron Paramagnetic Resonance of Nitroxide Spin-Labels at the Lower Stem, the Loop, and the Bulge
Y. Sun, Z. Zhang, V.M. Grigoryants, W.K. Myers, F. Liu, and K. Earle, J.H. Freed, C.P. Scholes
Biochemistry 51, 8530-8541 (2012).
Supporting Information
<doi: 10.1021/bi301058q>
PMID:
23009298
PMCID:
PMC3549007
|
|
|
ABSTRACT: Electron paramagnetic resonance (EPR) at 236.6 and 9.5 GHz probed the tumbling of nitroxide spin probes in the lower stem, in the upper loop, and near the bulge of mini c TAR DNA. High-frequency 236.6 GHz EPR, not previously applied to spin-labeled oligonucleotides, was notably sensitive to fast, anisotropic, hindered local rotational motion of the spin probe, occurring approximately about the NO nitroxide axis. Labels attached to the 2′-aminocytidine sugar in the mini c TAR DNA showed such anisotropic motion, which was faster in the lower stem, a region previously thought to be partially melted. More flexible labels attached to phosphorothioates at the end of the lower stem tumbled isotropically in mini c TAR DNA, mini TAR RNA, and ψ3 RNA, but at 5 °C, the motion became more anisotropic for the labeled RNAs, implying more order within the RNA lower stems. As observed by 9.5 GHz EPR, the slowing of nanosecond motions of large segments of the oligonucleotide was enhanced by increasing the ratio of the nucleocapsid protein NCp7 to mini c TAR DNA from 0 to 2. The slowing was most significant at labels in the loop and near the bulge. At a 4:1 ratio of NCp7 to mini c TAR DNA, all labels reported tumbling times of >5 ns, indicating a condensation of NCp7 and TAR DNA. At the 4:1 ratio, pulse dipolar EPR spectroscopy of bilabels attached near the 3′ and 5′ termini showed evidence of an NCp7-induced increase in the 3′–5′ end-to-end distance distribution and a partially melted stem.
|
|
|
Spin Labels in the Gel Phase and Frozen Lipid Bilayers: Do They Truly Manifest a Polarity Gradient?
B. Dzikowski and J.H. Freed.
In Nitroxides-Theory, Experiment and Applications; A. Kokorin. Ed.
InTech: Rijeka, Croatia, 2012; pp. 167-190, Ch. 5.
<doi: 10.5772/45769>
PMID: [None - Book] PMCID:
[None - Book]
|

|
|
INTRODUCTION: Lipid spin labels containing nitroxide groups at different positions in the fatty acid chain, such as 1-palmitoyl-2-stearoyl-(n-doxyl)-sn-glycero-3-phosphocholines (n-PC spin labels) are a useful and proven tool in lipid research. They have provided important insights into the structure of model and biological membranes, reported on the membrane fluidity, polarity, phase state and presence of microscopic domains, accessibility of different depth positions in the lipid bilayer for oxygen and other polar and non-polar paramagnetic compounds and protein/lipid interactions.
It is generally accepted, that, unlike bulky fluorescent labels, nitroxides are well incorporated into fluid lipid bilayers and not excluded from them. However, it has been shown by NMR that although the most probable location of the nitroxide group for 5-, 10- and 16- PC spin labels in the fluid POPC membrane corresponds to the fully extended conformation, the distribution is relatively broad and other conformations should also be present. Bent conformations were previously found for doxylstearic acids in monomolecular films, water/hydrocarbon emulsion particles and micellar systems. In fluid membranes the fluidity, polarity and accessibility parameters reported by ESR using PC spin labels and n-doxylstearic acids are, in general, change monotonically with an increase in n, although there are indications that the spin label groups on the stearates are located nearer to the membrane exterior than the analogous positions of the unlabeled phospholipid chains. However, in the gel phase, which is characterized by denser chain packing and higher order, the preferential location may be different.
In this chapter we focus on the behavior of PC spin labels in the gel phase and frozen membranes. We show how the superior g-factor resolution of HF ESR provides new insights in this behavior and a new look at the vast body of experimental data accumulated with PC spin labels in the last 30 years. In particular, we revisit so-called "polarity profiles" determined from the g-factor values and hyperfine splittings of PC spin labels in frozen phospholipid membranes with or without cholesterol and show that these values are affected by a number of factors in the membrane composition, chain packing in the lipid phase and folding properties of the sn-2 spin labeled chain PC labels rather than reflect gradients of polarity or water content present in the membrane.
|
|
|
Pulsed ESR Dipolar Spectroscopy for Distance Measurements in Immobilized Spin Labeled Proteins in Liquid Solution
Z. Yang, Y. Liu, P.P. Borbat, J.L. Zweier, J.H. Freed, and W.L. Hubbell.
J. Am. Chem. Soc. 134, 9950-9952 (2012).
Supporting Information
<doi: 10.1021/ja303791p>
PMID:
22676043
PMCID:
PMC3409244
|

|
|
ABSTRACT: Pulsed electron spin resonance (ESR) dipolar spectroscopy (PDS) in combination with site-directed spin labeling is unique in providing nanometer-range distances and distributions in biological systems. To date, most of the pulsed ESR techniques require frozen solutions at cryogenic temperatures to reduce the rapid electron spin relaxation rate and to prevent averaging of electron–electron dipolar interaction due to the rapid molecular tumbling. To enable measurements in liquid solution, we are exploring a triarylmethyl (TAM)-based spin label with a relatively long relaxation time where the protein is immobilized by attachment to a solid support. In this preliminary study, TAM radicals were attached via disulfide linkages to substituted cysteine residues at positions 65 and 80 or 65 and 76 in T4 lysozyme immobilized on Sepharose. Interspin distances determined using double quantum coherence (DQC) in solution are close to those expected from models, and the narrow distance distribution in each case indicates that the TAM-based spin label is relatively localized.
|
|
|
Electron Tunneling in Lithium-Ammonia Solutions Probed by Frequency-Dependent Electron Spin Relaxation Studies
K. Maeda, M.T.J. Lodge, J. Harmer, J.H. Freed, and P.P. Edwards.
J. Am. Chem. Soc. 134, 9209-18 (2012).
<doi: 10.1021/ja212015b>
PMID:
22568866
PMCID:
PMC3415590
|

|
|
ABSTRACT: Electron transfer or quantum tunneling dynamics for excess or solvated electrons in dilute lithium–ammonia solutions have been studied by pulse electron paramagnetic resonance (EPR) spectroscopy at both X- (9.7 GHz) and W-band (94 GHz) frequencies. The electron spin–lattice (T1) and spin–spin (T2) relaxation data indicate an extremely fast transfer or quantum tunneling rate of the solvated electron in these solutions which serves to modulate the hyperfine (Fermi-contact) interaction with nitrogen nuclei in the solvation shells of ammonia molecules surrounding the localized, solvated electron. The donor and acceptor states of the solvated electron in these solutions are the initial and final electron solvation sites found before, and after, the transfer or tunneling process. To interpret and model our electron spin relaxation data from the two observation EPR frequencies requires a consideration of a multiexponential correlation function. The electron transfer or tunneling process that we monitor through the correlation time of the nitrogen Fermi-contact interaction has a time scale of (1–10) × 10–12 s over a temperature range 230–290 K in our most dilute solution of lithium in ammonia. Two types of electron–solvent interaction mechanisms are proposed to account for our experimental findings. The dominant electron spin relaxation mechanism results from an electron tunneling process characterized by a variable donor–acceptor distance or range (consistent with such a rapidly fluctuating liquid structure) in which the solvent shell that ultimately accepts the transferring electron is formed from random, thermal fluctuations of the liquid structure in, and around, a natural hole or Bjerrum-like defect vacancy in the liquid. Following transfer and capture of the tunneling electron, further solvent-cage relaxation with a time scale of ˜10–13 s results in a minor contribution to the electron spin relaxation times. This investigation illustrates the great potential of multifrequency EPR measurements to interrogate the microscopic nature and dynamics of ultrafast electron transfer or quantum-tunneling processes in liquids. Our results also impact on the universal issue of the role of a host solvent (or host matrix, e.g. a semiconductor) in mediating long-range electron transfer processes and we discuss the implications of our results with a range of other materials and systems exhibiting the phenomenon of electron transfer.
|
|
|
Self-Association of the Histidine Kinase CheA as Studied by Pulsed Dipolar ESR Spectroscopy
J. Bhatnagar, R. Sircar, P.P. Borbat, J.H. Freed, and B.R. Crane.
Biophys. J. 102, 2192-2201 (2012).
Supporting Information
<doi: 10.1016/j.bpj.2012.03.038>
PMID:
22824284
PMCID:
PMC3341552
|

|
|
ABSTRACT: Biologically important protein complexes often involve molecular interactions that are low affinity or transient. We apply pulsed dipolar electron spin resonance spectroscopy and site-directed spin labeling in what to our knowledge is a new approach to study aggregation and to identify regions on protein surfaces that participate in weak, but specific molecular interactions. As a test case, we have probed the self-association of the chemotaxis kinase CheA, which forms signaling clusters with chemoreceptors and the coupling protein CheW at the poles of bacterial cells. By measuring the intermolecular dipolar interactions sensed by spin-labels distributed over the protein surface, we show that the soluble CheA kinase aggregates to a small extent through interactions mediated by its regulatory (P5) domain. Direct dipolar distance measurements confirm that a hydrophobic surface at the periphery of P5 subdomain 2 associates CheA dimers in solution. This result is further supported by differential disulfide cross-linking from engineered cysteine reporter sites. We suggest that the periphery of P5 is an interaction site on CheA for other similar hydrophobic surfaces and plays an important role in structuring the signaling particle.
|
|
|
Dynamics and Ordering of Lipid Spin-Labels Along the Coexistence Curve of Two Membrane Phases: An ESR Study
A.K. Smith and J.H. Freed.
Chem. Phys. Lipids, 165, 348-361 (2012).
Supporting Information
<doi: 10.1016/j.chemphyslip.2012.02.009>
PMID:
22586732
PMCID:
PMC3526980
|

|
|
ABSTRACT: An analysis of electron spin resonance (ESR) spectra from compositions along the liquid-ordered (Lo) and liquid-disordered (Ld) coexistence curve from the brain-sphingomyelin/dioleoylphosphatidylcholine/cholesterol (SPM/DOPC/Chol) model lipid system was performed to characterize the dynamic structure on a molecular level of these coexisting phases. We obtained 200 continuous-wave ESR spectra from glycerophospholipid spin-labels labeled at the 5, 7, 10, 12, 14, and 16 carbon positions of the 2nd acyl chain, a sphingomyelin spin-label labeled at the 14 carbon position of the amide-linked acyl chain, a headgroup-labeled glycerophospholipid, a headgroup-labeled sphingomyelin, and the cholesterol analogue spin-label cholestane all within multi-lamellar vesicle suspensions at room temperature. The spectra were analyzed using the MOMD (microscopic-order macroscopic-disorder) model to provide the rotational diffusion rates and order parameters which characterize the local molecular dynamics in these phases. The analysis also incorporated the known critical point and invariant points of the neighboring three-phase triangle along the coexistence curve. The variation in the molecular dynamic structures of coexisting Lo and Ld compositions as one moves toward the critical point is discussed. Based on these results, a molecular model of the Lo phase is proposed incorporating the "condensing effect" of cholesterol on the phospholipid acyl chain dynamics and ordering and the "umbrella model" of the phospholipid headgroup dynamics and ordering.
|
|
|
Effect of Freezing Conditions on Distances and Their Distributions Derived From Double Electron Electron Resonance (DEER): A Study of Doubly-Spin-Labeled T4 Lysozyme
E.R. Georgieva, A.S. Roy, V.M. Grigoryants, P.P. Borbat, K.A. Earle, C.P. Scholes, and J.H. Freed.
J. Magn. Reson. 216, 69-77 (2012).
Supporting Information
<doi: 10.1016/j.jmr.2012.01.004>
PMID:
22341208
PMCID:
PMC3323113
|

|
|
ABSTRACT: Pulsed dipolar ESR spectroscopy, DEER and DQC, require frozen samples. An important issue in the biological application of this technique is how the freezing rate and concentration of cryoprotectant could possibly affect the conformation of biomacromolecule and/or spin-label. We studied in detail the effect of these experimental variables on the distance distributions obtained by DEER from a series of doubly spin-labeled T4 lysozyme mutants. We found that the rate of sample freezing affects mainly the ensemble of spin-label rotamers, but the distance maxima remain essentially unchanged. This suggests that proteins frozen in a regular manner in liquid nitrogen faithfully maintain the distance-dependent structural properties in solution.
We compared the results from rapidly freeze-quenched (≤100 μs) samples to those from commonly shock-frozen (slow freeze, 1 s or longer) samples. For all the mutants studied we obtained inter-spin distance distributions, which were broader for rapidly frozen samples than for slowly frozen ones. We infer that rapid freezing trapped a larger ensemble of spin label rotamers; whereas, on the time-scale of slower freezing the protein and spin-label achieve a population showing fewer low-energy conformers. We used glycerol as a cryoprotectant in concentrations of 10% and 30% by weight. With 10% glycerol and slow freezing, we observed an increased slope of background signals, which in DEER is related to increased local spin concentration, in this case due to insufficient solvent vitrification, and therefore protein aggregation. This effect was considerably suppressed in slowly frozen samples containing 30% glycerol and rapidly frozen samples containing 10% glycerol. The assignment of bimodal distributions to tether rotamers as opposed to protein conformations is aided by comparing results using MTSL and 4-Bromo MTSL spin-labels. The latter usually produce narrower distance distributions.
|
|
|
Conformational Distributions and Hydrogen Bonding in Gel and Frozen Lipid Bilayers: A High Frequency Spin-Label ESR Study
B. Dzikovski, D. Tipikin, and J. Freed
J. Phys. Chem. B, 116, 6694-6706 (2012).
Supporting Information
<doi: 10.1021/jp211879s>
PMID:
22324811
PMCID:
PMC3376253
|
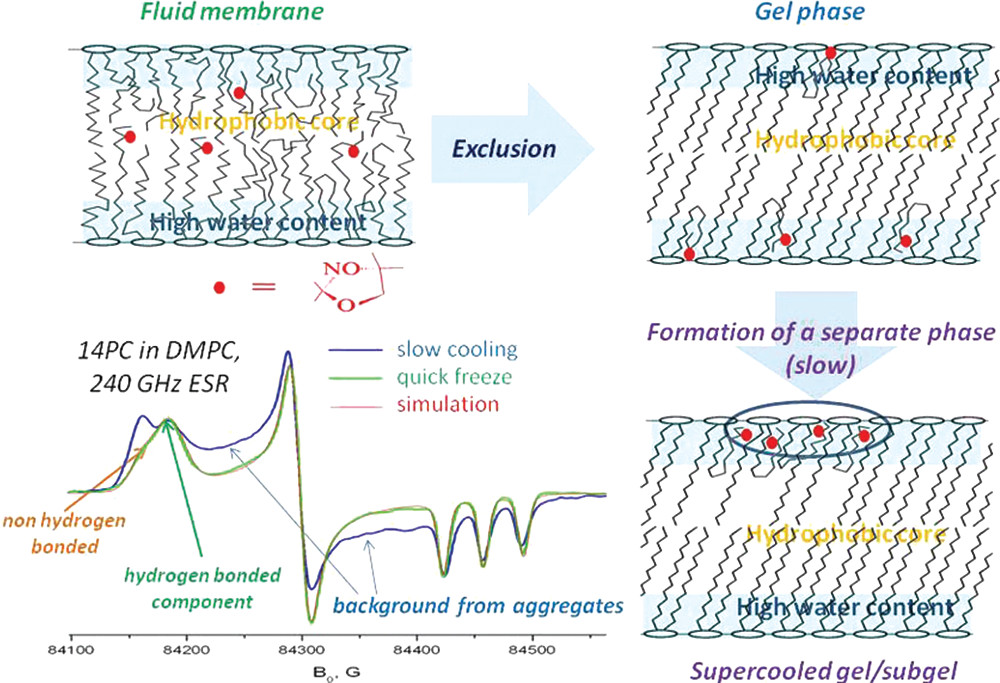
|
|
ABSTRACT: The ESR parameters of PC spin labels in frozen membranes do not simply represent the membrane polarity or water penetration profile. Instead, they show a distribution between hydrogen-bonded (HB) and non-hydrogen-bonded (non-HB) states, which is affected by a number of factors in the membrane composition. Similar to the exclusion of solutes from crystallizing solvents, the pure bulk gel phase excludes nitroxides, forcing acyl chains to take bent conformations. In these conformations, the nitroxide is hydrogen-bonded. Furthermore, upon gradual cooling in the supercooled gel, PC labels undergo slow lateral aggregation, resulting in a broad background signal. However, if the sample is instantly frozen, this background is replaced by the HB component. In membranes with cholesterol, the observed HB/non-HB ratio can best be described by a partition-like equilibrium between nitroxides located in defects of lipid structure within the hydrophobic core and those close to the membrane surface.
|
|
|
A Protocol for Detecting and Scavenging Gas-phase Free Radicals in Mainstream Cigarette Smoke
L.X. Yu, B.G. Dzikovski, and J.H. Freed.
J. Vis. Exp. 59, e3406 (2012).
<doi: 10.3791/3406>
PMID:
22230844
PMCID:
PMC3369772
|

|
|
SUMMARY: Spin-trapping ESR spectroscopy was used to study the effect of plant antioxidants lycopene, pycnogenol and grape seed extract on scavenging gas-phase free radicals in cigarette smoke.
ABSTRACT: Cigarette smoking is associated with human cancers. It has been reported that most of the lung cancer deaths are caused by cigarette smoking. Although tobacco tars and related products in the particle phase of cigarette smoke are major causes of carcinogenic and mutagenic related diseases, cigarette smoke contains significant amounts of free radicals that are also considered as an important group of carcinogens. Free radicals attack cell constituents by damaging protein structure, lipids and DNA sequences and increase the risks of developing various types of cancers. Inhaled radicals produce adducts that contribute to many of the negative health effects of tobacco smoke in the lung. Studies have been conducted to reduce free radicals in cigarette smoke to decrease risks of the smoking-induced damage. It has been reported that haemoglobin and heme-containing compounds could partially scavenge nitric oxide, reactive oxidants and carcinogenic volatile nitrosocompounds of cigarette smoke. A 'bio-filter' consisted of haemoglobin and activated carbon was used to scavenge the free radicals and to remove up to 90% of the free radicals from cigarette smoke. However, due to the cost-ineffectiveness, it has not been successfully commercialized. Another study showed good scavenging efficiency of shikonin, a component of Chinese herbal medicine. In the present study, we report a protocol for introducing common natural antioxidant extracts into the cigarette filter for scavenging gas phase free radicals in cigarette smoke and measurement of the scavenge effect on gas phase free radicals in mainstream cigarette smoke (MCS) using spin-trapping Electron Spin Resonance (ESR) Spectroscopy. We showed high scavenging capacity of lycopene and grape seed extract which could point to their future application in cigarette filters. An important advantage of these prospective scavengers is that they can be obtained in large quantities from byproducts of tomato or wine industry respectively.
|
|
|
Protein Dynamics by NMR Spin Relaxation: The Slowly Relaxing Local Structure Perspective
E. Meirovitch, A. Polimeno, and J.H. Freed.
In E. Mag. Res. Online, R.K. Harris and R.L. Wasylishen, Eds.
John Wiley & Sons, Inc.: New York, 2011; pp 1-9.
<doi: 10.1002/9780470034590.emrstm1243>
PMID: [None - Book] PMCID:
[None - Book]
|
|
|
ABSTRACT: The two-body (protein and probe) coupled rotator slowly relaxing local structure (SRLS) approach, implemented for NMR spin relaxation in proteins, is presented. SRLS is based on a Smoluchowki equation that includes the tensorial descriptions of the diffusing rotators and (via the local ordering) their dynamical coupling. This yields spectral densities that enter the expressions for the experimentally measured relaxation parameters. The constrained motion of the probe is treated by analogy with the standard treatments of restricted motions in liquids. Such restricted motions are largely recovered in the limit of large time scale separation between the rotators, i.e., when mode-coupling is not important. We found that the tensorial properties are not simple for proteins even in this limit. Their complexity has to be accounted for to obtain physically insightful pictures of protein dynamics. The traditional method for analyzing NMR spin relaxation in proteins is model-free (MF). This method treats only the simplest tensorial properties and implies large time-scale separation. Only when these conditions are fulfilled the analytical MF spectral densities, which are limiting cases of SRLS, may be used. Since they are not fulfilled in most actual cases, the MF-based pictures of protein dynamics are unreliable.
|
|
|
Stochastic Methods for Magnetic Resonance Spectroscopies
A. Polimeno, V. Barone, and J.H. Freed.
In Computational Strategies for Spectroscopy: From Small Molecules to Nano Systems; V. Barone, Ed.
Wiley: New York, NY, 2012; Chapter 12.
<doi: 10.1002/9781118008720.ch12>
PMID: [None - Book] PMCID:
[None - Book]
|

|
|
ABSTRACT: Physicochemical properties of molecules in solution depend on the action of different motions at several time and length scales, and information on multiscale dynamics can be gained, in principle, by a variety of spectroscopic techniques. In this work we review theoretical tools for the investigation of "slow" molecular motions, such as solvent cage effects in liquids and liquid crystals, global and local dynamics in proteins, reorientation dynamics, and internal (conformational) degrees of freedom. Spectroscopic techniques which are most sensitive to such motions are electron spin resonance and nuclear magnetic resonance and they require ad hoc theoretical treatment. In particular, we discuss the definition of multidimensional stochastic models and their treatment to interpret magnetic resonance spectroscopic data of rigid and flexible molecules in isotropic media, liquid crystals, and biosystems.
|
|
|
ESR Microscopy for Biological and Biomedical Applications
C.S. Shin, C.R. Dunnam, P.P. Borbat, B. Dzikovski, E.D. Barth, H.J. Halpern, and J.H. Freed.
Nanoscience & Nanotechnology Letters 3, 561-567 (2011).
<doi: 10.1166/nnl.2011.1206>
PMID:
21984955
PMCID:
PMC3188420
|

|
|
ABSTRACT: We report on electron-spin resonance microscopy (ESRM) providing sub-micron resolution (˜700nm) with a high spin concentration sample, i.e. lithium phthalocyanine (LiPc) crystal. For biomedical applications of our ESRM, we have imaged samples containing rat basophilic leukemia (RBL) cells as well as cancerous tissue samples with a resolution of several microns using a water soluble spin probe, Trityl_OX063_d24. Phantom samples with the nitroxide spin label, 15N PDT, were also imaged to demonstrate that nitroxides, which are commonly used as spin labels, may also be used for ESRM applications. ESRM tissue imaging would therefore be valuable for diagnostic or therapeutic purposes. Also, ESRM can be used to study the motility or the metabolism of cells in various environments. With further modification and/or improvement of imaging probe and spectrometer instrumentation sub-micron biological images should be obtainable, thereby providing a useful tool for various biomedical applications.
|
|
|
2D-ELDOR Study of Heterogeneity and Domain Structure Changes in Plasma Membrane Vesicles upon Cross-Linking of Receptors
Y.-W. Chiang, A.J. Costa-Filho, B. Baird, and J.H. Freed.
J. Phys. Chem. B 115, 10462-10469 (2011).
Supporting Information
<doi: 10.1021/jp2016243>
PMID:
21780815
PMCID:
PMC3165081
|
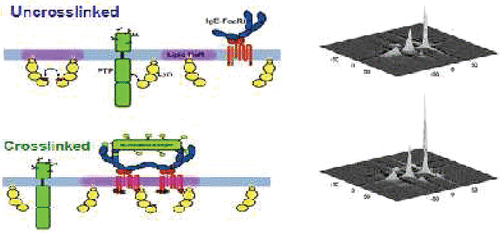
|
|
ABSTRACT: 2D electron–electron double resonance (2D-ELDOR) with the "full Sc–" method of analysis is applied to the study of plasma membrane vesicles. Membrane structural changes upon antigen cross-linking of IgE receptors (IgE-FcεRI) in plasma membrane vesicles (PMVs) isolated from RBL-2H3 mast cells are investigated, for the first time, by means of these 2D-ELDOR techniques. Spectra of 1-palmitoyl-2-(16-doxyl stearoyl) phosphatidylcholine (16-PC) from PMVs before and after this stimulation at several temperatures are reported. The results demonstrate a coexistence of liquid-ordered (Lo) and liquid-disordered (Ld) components. We find that upon cross-linking, the membrane environment is remodeled to become more disordered, as shown by a moderate increase in the population of the Ld component. This change in the relative amount of the Lo versus Ld components upon cross-linking is consistent with a model wherein the IgE receptors, which when clustered by antigen to cause cell stimulation, lead to more disordered lipids, and their dynamic and structural properties are slightly altered. This study demonstrates that 2D-ELDOR, analyzed by the full Sc– method, is a powerful approach for capturing the molecular dynamics in biological membranes. This is a particular case showing how 2D-ELDOR can be applied to study physical processes in complex systems that yield subtle changes.
|
|
|
Distance Measurements: Continuous-Wave (CW)- and Pulsed Dipolar EPR
S.K. Misra and J.H. Freed.
In Multifrequency Electron Paramagnetic Resonance, S.K. Misra, Ed.
Wiley-VCH: New York, 2011; Chapter 12, pp. 545-588.
<doi: 10.1002/9783527633531.ch12>
PMID: [None - Book] PMCID:
[None - Book]
|
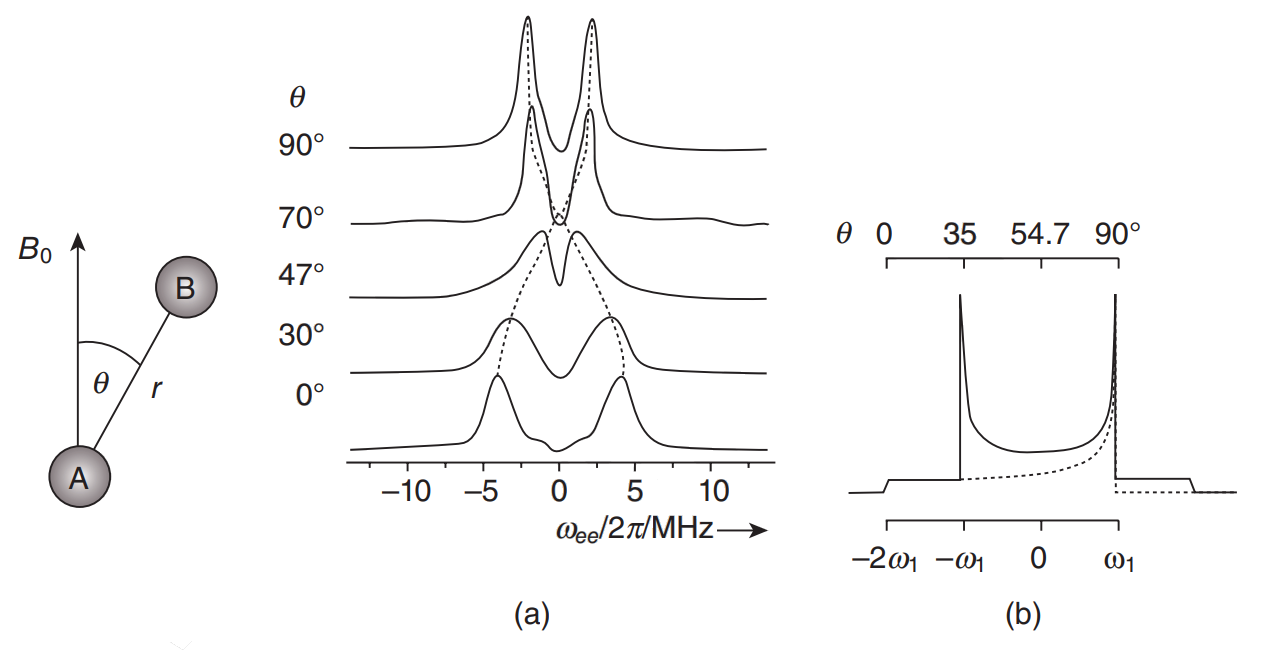
|
|
INTRODUCTION: Information on long-range distances between selected sites is a prerequisite to understanding the detailed structure of complex systems. In NMR utilizing a spin label, it is possible to measure distances in the range between 8 Å to, at most, 25 Å. On the other hand, with EPR, distances in the range between about 10 and 90 Å can and have been measured. EPR spectroscopy may be the best practical technique for complex systems, since methods such as small-angle scattering, X-ray scattering and small-angle neutron scattering have insufficient contrast whereas fluorescence resonant energy transfer (FRET) lacks key virtues of EPR noted below. Distance measurements in EPR are based on exploitation of the dipolar interaction (DI), that depends on the distance between two paramagnetic probes, and which may be identical or may differ from each other, and are located at different spatial positions in the sample. They can be introduced using site-directed mutagenesis (also known as site-directed spin labeling; SDSL), a technique which enables the investigation of, for example, proteins and other biomolecules. In fact, the signature of the DI can be clearly seen in CW-EPR spectra under favorable circumstances for short distances. On the other hand, in pulsed EPR [hereafter referred to as pulsed dipolar spectroscopy; PDS), experiments can be designed specifically to clearly distinguish the dipolar interaction. The two commonly used PDS techniques are: (i) pulsed electron double resonance (PELDOR; this term was originally coined by the Russians, who first developed the technique), which is also referred to as double electron-electron resonance (DEER; this term was introduced later in the USA, and will be used hereafter in this chapter); and (ii) double quantum coherence (DQC)-EPR.
|
|
|
Molecular Motions
S.K. Misra and J.H. Freed.
In Multifrequency Electron Paramagnetic Resonance, S.K. Misra, Ed.
Wiley-VCH: New York, 2011; Chapter 11, pp. 497-544.
|

|
|
Molecular Motions
S.K. Misra and J.H. Freed.
In Multifrequency Electron Paramagnetic Resonance, S.K. Misra, Ed.
Wiley-VCH: New York, 2011; Chapter 11, pp. 497-544.
<doi: 10.1002/9783527633531.ch11>
PMID: [None - Book] PMCID:
[None - Book]
|
|
|
INTRODUCTION: In this chapter we will discuss the study of molecular motion by EPR, using both continuous-wave (CW) and pulsed EPR, in particular two-dimensional electron–electron double resonance (2-D-ELDOR). In recent years, the study of protein dynamics using site-directed spin labels (SDSL) by EPR has become an important subject.
|
|
|
A New Lanczos-Based Algorithm for Simulating High-Frequency Two-Dimensional Electron Spin Resonance Spectra
Y.-W. Chiang and J.H. Freed.
J. Chem. Phys. 134, 034112 (2011).
<doi: 10.1063/1.3523576>
PMID:
21261335
PMCID:
PMC3041155
|
|
|
ABSTRACT: The Lanczos algorithm (LA) is a useful iterative method for the reduction of a large matrix to tridiagonal form. It is a storage efficient procedure requiring only the preceding two Lanczos vectors to compute the next. The quasi-minimal residual (QMR) method is a powerful method for the solution of linear equation systems, Ax = b. In this report we provide another application of the QMR method: we incorporate QMR into the LA to monitor the convergence of the Lanczos projections in the reduction of large sparse matrices. We demonstrate that the combined approach of the LA and QMR can be utilized efficiently for the orthogonal transformation of large, but sparse, complex, symmetric matrices, such as are encountered in the simulation of slow-motional 1D- and 2D-electron spin resonance (ESR) spectra. Especially in the 2D-ESR simulations, it is essential that we store all of the Lanczos vectors obtained in the course of the LA recursions and maintain their orthogonality. In the LA-QMR application, the QMR weight matrix mitigates the problem that the Lanczos vectors lose orthogonality after many LA projections. This enables substantially more Lanczos projections, as required to achieve convergence for the more challenging ESR simulations. It, therefore, provides better accuracy for the eigenvectors and the eigenvalues of the large sparse matrices originating in 2D-ESR simulations than does the previously employed method, which is a combined approach of the LA and the conjugate-gradient (CG) methods, as evidenced by the quality and convergence of the 2D-ESR simulations. Our results show that very slow-motional 2D-ESR spectra at W-band (95 GHz) can be reliably simulated using the LA-QMR method, whereas the LA-CG consistently fails. The improvements due to the LA-QMR are of critical importance in enabling the simulation of high-frequency 2D-ESR spectra, which are characterized by their very high resolution to molecular orientation.
|
|
|
Joint Analysis of ESR Lineshapes and 1H NMRD Profiles of DOTA-Gd Derivatives by Means of the Slow Motion Theory
D. Kruk, J. Kowalewski, D.S. Tipikin, J.H. Freed, M. Mościcki, A. Mielczarek, and M. Port.
J. Chem. Phys. 134, 024508 (2011).
<doi: 10.1063/1.3516590>
PMID:
21241121
PMCID:
PMC3188623
|
|
|
ABSTRACT: The "Swedish slow motion theory" applied so far to Nuclear Magnetic Relaxation Dispersion (NMRD) profiles for solutions of transition metal ion complexes has been extended to ESR spectral analysis, including in addition g-tensor anisotropy effects. The extended theory has been applied to interpret in a consistent way (within one set of parameters) NMRD profiles and ESR spectra at 95 and 237 GHz for two Gd(III) complexes denoted as P760 and P792 (hydrophilic derivatives of DOTA-Gd, with molecular masses of 5.6 and 6.5 kDa, respectively). The goal is to verify the applicability of the commonly used pseudorotational model of the transient zero field splitting (ZFS). According to this model the transient ZFS is described by a tensor of a constant amplitude, defined in its own principal axes system, which changes its orientation with respect to the laboratory frame according to the isotropic diffusion equation with a characteristic time constant (correlation time) reflecting the time scale of the distortional motion. This unified interpretation of the ESR and NMRD leads to reasonable agreement with the experimental data, indicating that the pseudorotational model indeed captures the essential features of the electron spin dynamics.
|
|
|
Two Conserved Residues Are Important for Inducing Highly Ordered Membrane Domains by the Transmembrane Domain of Influenza Hemagglutinin
M. Ge and J.H. Freed.
Biophys. J. 100, 90-97 (2011).
Supporting Information
<doi: 10.1016/j.bpj.2010.11.014>
PMID:
21190660
PMCID:
PMC3010018
|

|
|
ABSTRACT: The interaction with lipids of a synthetic peptide corresponding to the transmembrane domain of influenza hemagglutinin was investigated by means of electron spin resonance. A detailed analysis of the electron spin resonance spectra from spin-labeled phospholipids revealed that the major effect of the peptide on the dynamic membrane structure is to induce highly ordered membrane domains that are associated with electrostatic interactions between the peptide and negatively charged lipids. Two highly conserved residues in the peptide were identified as being important for the membrane ordering effect. Aggregation of large unilamellar vesicles induced by the peptide was also found to be correlated with the membrane ordering effect of the peptide, indicating that an increase in membrane ordering, i.e., membrane dehydration, is important for vesicle aggregation. The possibility that hydrophobic interaction between the highly ordered membrane domains plays a role in vesicle aggregation and viral fusion is discussed.
|
|
|
Methyl Dynamics of a Ca2+-Calmodulin-Peptide Complex from NMR/SRLS
Y.E. Shapiro, A. Polimeno, J.H. Freed, and E. Meirovitch.
J. Phys. Chem. B 115, 354-365 (2011).
<doi: 10.1021/jp107130m>
PMID:
21166433
PMCID:
PMC3062514
|

|
|
ABSTRACT: We developed the slowly relaxing local structure (SRLS) approach for analyzing NMR spin relaxation in proteins. SRLS accounts for dynamical coupling between the tumbling of the protein and the local motion of the probe and for general tensorial properties. It is the generalization of the traditional model-free (MF) method, which does not account for mode-coupling and treats only simple tensorial properties. SRLS is applied herein to 2H relaxation of 13CDH2 groups in the complex of Ca2+–calmodulin with the peptide smMLCKp. Literature data comprising 2H T1 and T2 acquired at 14.1 and 17.6 T, and 288, 295, 308, and 320 K, are used. We find that mode-coupling is a small effect for methyl dynamics. On the other hand, general tensorial properties are important. In particular, it is important to allow for the asymmetry of the local spatial restrictions, which can be represented in SRLS by a rhombic local ordering tensor with components S02 and S22. The principal axes frame of this tensor is obviously different from the axial frames of the magnetic tensors. Here, we find that −0.2 ≤ S02 ≤ 0.5 and −0.4 ≤ S22 ≤ 0. MF features a single "generalized" order parameter, S, confined to the 0−0.316 range; the local geometry is inherently simple. The parameter S is inaccurate, having absorbed unaccounted for effects, notably S22 ≠ 0. We find that the methionine methyls (the other methyl types) reorient with rates of 8.6 × 109 to 21.4 × 109 (0.67 × 109 to 6.5 × 109) 1/s. The corresponding activation energies are 10 (10−27) kJ/mol. By contrast, MF yields inaccurate effective local motional correlation times, τe, with nonphysical temperature dependence. Thus, the problematic S- and τe-based MF picture of methyl dynamics has been replaced with an insightful physical picture based on a local ordering tensor related to structural features, and a local diffusion tensor that yields accurate activation energies.
|
|
|
Backbone Dynamics of Deoxy and Carbonmonoxy Hemoglobin by NMR/SRLS
E. Meirovitch, M. Zerbetto, A. Polimeno, and J.H. Freed.
J. Phys. Chem. B 115, 143-157 (2011).
<doi: 10.1021/jp107553j>
PMID:
21162544
PMCID:
PMC3071157
|
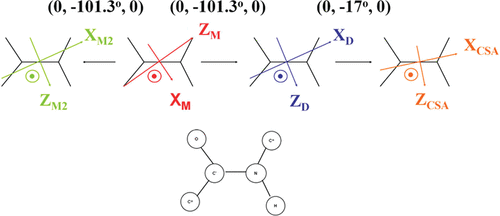
|
|
ABSTRACT: The slowly relaxing local structure (SRLS) approach, developed for NMR spin relaxation analysis in proteins, is applied herein to amide 15N relaxation in deoxy and carbonmonoxy hemoglobin. Experimental data including 15N T1, T2 and 15N-{1H} NOE, acquired at 11.7 and 14.1 T, and 29 and 34 °C, are analyzed. The restricted local motion of the N–H bond is described in terms of the principal value (S02) and orientation (βD) of an axial local ordering tensor, S, and the principal values (R∥L andR⊥L) and orientation (βO) of an axial local diffusion tensor, RL. The parameters c02 (the potential coefficient in terms of which S02 is defined), R∥L, βD, and βO are determined by data fitting; R⊥L is set equal to the global motional rate, RC, found previously to be (5.2–5.8) × 106 1/s in the temperature range investigated. The principal axis of S is (nearly) parallel to the Ci–1α–Ciα axis; when the two axes are parallel, βD = –101.3° (in the frame used). The principal axis of RL is (nearly) parallel to the N–H bond; when the two axes are parallel, βO = –101.3°. For "rigid" N–H bonds located in secondary structure elements the best-fit parameters are S02 = 0.88–0.95 (corresponding to local potentials of 8.6–19.9 kBT), R∥L = 109–1010 1/s, βD = –101.3° ± 2.0°, and βO = –101.3° ± 4°. For flexible N–H bonds located in loops the best-fit values are S02 = 0.75–0.80 (corresponding to local potentials of 4.5–5.5 kBT), R∥L = (1.0–6.3) × 108 1/s, βD = –101.3° ± 4.0°, and βO = –101.3° ± 10°. These results are important in view of their physical clarity, inherent potential for further interpretation, consistency, and new qualitative insights provided (vide infra).
|
|
|
Channel and Nonchannel Forms of Spin-Labeled Gramicidin in Membranes and Their Equilibria
B.G. Dzikovski, P.P. Borbat, and J.H. Freed.
J. Phys. Chem. B 115, 176-185 (2011).
Supporting Information
<doi: 10.1021/jp108105k>
PMID:
21142163
PMCID:
PMC3076037
|

|
|
ABSTRACT: Channel and nonchannel forms of gramicidin A (GA) were studied by ESR in various lipid environments using new mono- and double-spin-labeled compounds. For GA channels, we demonstrate here how pulse dipolar ESR can be used to determine the orientation of the membrane-traversing molecule relative to the membrane normal and to study subtle effects of lipid environment on the interspin distance in the spin-labeled gramicidin channel. To study nonchannel forms of gramicidin, pulse dipolar ESR was used first to determine interspin distances corresponding to monomers and double-helical dimers of spin-labeled GA molecules in the organic solvents trifluoroethanol and octanol. The same distances were then observed in membranes. Since detection of nonchannel forms in the membrane is complicated by aggregation, we suppressed any dipolar spectra from intermolecular interspin distances arising from the aggregates by using double-labeled GA in a mixture with excess unlabeled GA. In hydrophobic mismatching lipids (Lβ phase of DPPC), gramicidin channels dissociate into free monomers. The backbone structure of the monomeric form is similar to a monomeric unit of the channel dimer. In addition to channels and monomers, the double-helical conformation of gramicidin is present in some membrane environments. In the gel phase of saturated phosphatidylcholines, the fraction of double helices increases in the following order: DLPC < DMPC < DSPC < DPPC. The equilibrium DHD/monomer ratio in DPPC was determined. In membranes, the double-helical form is present only in aggregates. In addition, we studied the effect of N-terminal substitution in the GA molecule upon channel formation. This work demonstrates how pulsed dipolar ESR may be utilized to study complex equilibria of peptides in membranes.
|
|
|
Mechanistic Understanding of Pyrococcus horikoshii Dph2, a [4Fe-4S] Enzyme Required for Diphthamide Biosynthesis
X. Zhu, B. Dzikovski, X. Su, A.T. Torelli, Y. Zhang, S.E. Ealick, J.H. Freed and H. Lin.
Mol. Biosyst. 7, 74-81 (2011).
<doi: 10.1039/c0mb00076k>
PMID:
20931132
PMCID:
PMC3066188
|

|
|
ABSTRACT: Diphthamide, the target of diphtheria toxin, is a unique posttranslational modification on eukaryotic and archaeal translation elongation factor 2 (EF2). The proposed biosynthesis of diphthamide involves three steps and we have recently found that in Pyrococcus horikoshii (P. horikoshii), the first step uses an S-adenosyl-L-methionine (SAM)-dependent [4Fe–4S] enzyme, PhDph2, to catalyze the formation of a C–C bond. Crystal structure shows that PhDph2 is a homodimer and each monomer contains three conserved cysteine residues that can bind a [4Fe–4S] cluster. In the reduced state, the [4Fe–4S] cluster can provide one electron to reductively cleave the bound SAM molecule. However, different from classical radical SAM family of enzymes, biochemical evidence suggest that a 3-amino-3-carboxypropyl radical is generated in PhDph2. Here we present evidence supporting that the 3-amino-3-carboxypropyl radical does not undergo hydrogen abstraction reaction, which is observed for the deoxyadenosyl radical in classical radical SAM enzymes. Instead, the 3-amino-3-carboxypropyl radical is added to the imidazole ring in the pathway towards the formation of the product. Furthermore, our data suggest that the chemistry requires only one [4Fe–4S] cluster to be present in the PhDph2 dimer.
|
|
|
The Lipid-Binding Domain of Wild Type and Mutant α-Synuclein: Compactness and Interconversion Between the Broken and Extended Helix Forms
E.R. Georgieva, T.F. Ramlall, P.P. Borbat, J.H. Freed, and D. Eliezer.
J. Biol. Chem. 285, 28261-28274 (2010).
Supporting Information
<doi: 10.1074/jbc.M110.157214>
PMID:
20592036
PMCID:
PMC2934691
|

|
|
ABSTRACT: α-Synuclein (αS) is linked to Parkinson disease through its deposition in an amyloid fibril form within Lewy Body deposits, and by the existence of three αS point mutations that lead to early onset autosomal dominant Parkinsonism. The normal function of αS is thought to be linked to the ability of the protein to bind to the surface of synaptic vesicles. Upon binding to vesicles, αS undergoes a structural reorganization from a dynamic and disordered ensemble to a conformation consisting of a long extended helix. In the presence of small spheroidal detergent micelles, however, this extended helix conformation can convert into a broken helix state, in which a region near the middle of the helix unwinds to form a linker between the two resulting separated helices. Membrane-bound conformations of αS likely mediate the function of the protein, but may also play a role in the aggregation and toxicity of the protein. Here we have undertaken a study of the effects of the three known PD-linked mutations on the detergent- and membrane-bound conformations of αS, as well as factors that govern the transition of the protein between the extended helix and broken helix states. Using pulsed dipolar ESR measurements of distances up to 8.7 nm, we show that all three PD-linked αS mutants retain the ability to transition from the broken helix to the extended helix conformation. In addition, we find that the ratio of protein to detergent, rather than just the absolute detergent concentration, determines whether the protein adopts the broken or extended helix conformation.
|
|
|
A Multifrequency EPR Study of Fe2+ and Mn2+ Ions in a ZnSiF6·6H2O Single Crystal at Liquid-Helium Temperatures
S.K. Misra, S. Diehl, D. Tipikin, J.H. Freed.
J. Magn. Res. 205, 14-22 (2010).
<doi: 10.1016/j.jmr.2010.03.013>
PMID:
20395160
PMCID:
PMC2885525
|

|
|
ABSTRACT: A liquid-helium temperature study of Fe2+ and Mn2+ ions has been carried out on a single crystal of Fe2+-doped ZnSiF6⋅6H2O at 5–35 K at 170, 222.4 and 333.2 GHz. The spectra are found to be an overlap of two magnetically inequivalent Fe2+ ions, as well as that of an Mn2+ ion. From the simulation of the EPR line positions for the Fe2+ (d6, S = 2) ion the spin-Hamiltonian parameters were estimated for the two inequivalent Fe2+ ions at the various temperatures. From the relative intensities of lines the absolute sign of the fine-structure parameters have been estimated. In addition, the fine-structure and hyperfine-structure spin-Hamiltonian parameters for the Mn2+ ion, present as impurity at interstitial sites, were estimated from the hyperfine allowed and forbidden line positions. The particular virtues of such a single-crystal study vs. that on powders are noted.
|
|
|
Diphthamide Biosynthesis Requires an Organic Radical Generated by an Iron-Sulphur Enzyme
Y. Zhang, X. Zhu, A.T. Torelli, M. Lee, B. Dzikovski, R.M. Koralewski, E. Wang, J.H. Freed, C. Krebs, S.E. Ealick, and H. Lin.
Nature, 465, 891-896 (2010).
|

|
|
Diphthamide Biosynthesis Requires an Organic Radical Generated by an Iron-Sulphur Enzyme
Y. Zhang, X. Zhu, A.T. Torelli, M. Lee, B. Dzikovski, R.M. Koralewski, E. Wang, J.H. Freed, C. Krebs, S.E. Ealick, and H. Lin.
Nature, 465, 891-896 (2010).
Supporting Information
<doi: 10.1038/nature09138>
PMID:
20559380
PMCID:
PMC3006227
|
|
|
ABSTRACT: Archaeal and eukaryotic translation elongation factor 2 contain a unique post-translationally modified histidine residue called diphthamide, which is the target of diphtheria toxin. The biosynthesis of diphthamide was proposed to involve three steps, with the first being the formation of a C–C bond between the histidine residue and the 3-amino-3-carboxypropyl group of S-adenosyl-L-methionine (SAM). However, further details of the biosynthesis remain unknown. Here we present structural and biochemical evidence showing that the first step of diphthamide biosynthesis in the archaeon Pyrococcus horikoshii uses a novel iron–sulphur-cluster enzyme, Dph2. Dph2 is a homodimer and each of its monomers can bind a [4Fe–4S] cluster. Biochemical data suggest that unlike the enzymes in the radical SAM superfamily, Dph2 does not form the canonical 5′-deoxyadenosyl radical. Instead, it breaks the Cγ,Met–S bond of SAM and generates a 3-amino-3-carboxypropyl radical. Our results suggest that P. horikoshii Dph2 represents a previously unknown, SAM-dependent, [4Fe–4S]-containing enzyme that catalyses unprecedented chemistry.
|
|
|
Comment on "The physical basis of model-free analysis of NMR relaxation data from proteins and complex fluids" [J. Chem. Phys. 131, 224507 (2009)]
E. Meirovitch, A. Polimeno, and J.H. Freed.
J. Chem. Phys. 132, 207101 (2010).
<doi: 10.1063/1.3429599>
PMID:
20515116
PMCID:
PMC2887917
|
|
|
In a recent article, Halle was critical of the use of the slowly relaxing local structure (SRLS) model for the analysis of NMR spin relaxation in proteins. Although he cited private communications with us, he did not discuss the contents of his article prior to its publication, nor were we aware of his intentions. We are in disagreement with a number of aspects of this article, but confine ourselves just to a brief rebuttal of his negative comments about SRLS and its use. We do, however, respect Halle’s efforts to provide a physical basis for the model-free (MF) approach.
|
|
|
Structural Dynamics of Bio-Macromolecules by NMR: The Slowly Relaxing Local Structure Approach
E. Meirovitch, Y.E. Shapiro, A. Polimeno, J.H. Freed.
Progress in NMR Spectroscopy, 56, 360-405 (2010).
<doi: 10.1016/j.pnmrs.2010.03.002>
PMID:
20625480
PMCID:
PMC2899824
|

|
|
INTRODUCTION: Protein dynamics by NMR has been reviewed extensively in recent years. These surveys show decisively that information on structure should be complemented by information on motion both to properly characterize the protein, and to understand its function. The time scale accessible by NMR extends from picoseconds to days, with different methods accessing different parts of this time axis. Here we focus on heteronuclear NMR spin relaxation used to study ps to ns protein dynamics. The slow limit of this time regime is determined by the global tumbling of the protein, with the rates for internal motion of the probe being typically faster.
|
|
|
Multifrequency Electron Spin Resonance Study of the Dynamics of Spin Labeled T4 Lysozyme
Z. Zhang, M.R. Fleissner, D.S. Tipikin, Z. Liang, J.K. Moscicki, K.A. Earle, W.L. Hubbell, and J.H. Freed.
J. Phys. Chem. B, 114, 5503-5521 (2010).
Supporting Information
<doi: 10.1021/jp910606h>
PMID:
20361789
PMCID:
PMC2865245
|

|
|
ABSTRACT: An extensive set of electron spin resonance spectra was obtained over a wide range of frequencies (9, 95, 170, and 240 GHz) and temperatures (2 to 32 °C) to explore the dynamic modes of nitroxide-labeled T4 lysozyme in solution. A commonly used nitroxide side chain (R1), or a methylated analogue with hindered internal motion (R2), was substituted for the native side chain at solvent-exposed helical sites, 72 or 131. The spectra at all four frequencies were simultaneously fit with the slowly relaxing local structure (SRLS) model. Good fits were achieved at all the temperatures. Two principle dynamic modes are included in the SRLS model, the global tumbling of the protein and the internal motion consisting of backbone fluctuations and side chain isomerizations. Three distinct spectral components were required for R1 and two for R2 to account for the spectra at all temperatures. One is a highly ordered and slow motional component, which is observed in the spectra of both R1 and R2; it may correspond to conformers stabilized by interaction with the protein surface. The fraction of this component decreases with increasing temperature and is more populated in the R2 spectra, possibly arising from stronger interaction of the nitroxide ring with the protein surface due to the additional methyl group. The other two components of R1 and the second component of R2 are characterized by fast anisotropic diffusion and relatively low ordering, most likely corresponding to conformers having little or no interactions with nearby residues. Ficoll of different concentrations was added to increase the solution viscosity, thereby slowing down the global tumbling of the protein. A significant effect of Ficoll on the internal motion of an immobilized component was apparent in R2 but not in R1. The ability of such multifrequency studies to separate the effects of faster internal modes of motion from slower overall motions is clearly demonstrated, and its utility in future studies is considered.
|
|
|
Structure of the Ternary Complex Formed by a Chemotaxis Receptor Signaling Domain, the CheA Histidine Kinase, and the Coupling Protein CheW As Determined by Pulsed Dipolar ESR Spectroscopy
J. Bhatnagar, P.P. Borbat, A.M. Pollard, A.M. Bilwes, J.H. Freed, and B.R. Crane.
Biochemistry, 49, 3824-3841 (2010).
Supporting Information
<doi: 10.1021/bi100055m>
PMID:
20355710
PMCID:
PMC2873776
|

|
|
ABSTRACT: The signaling apparatus that controls bacterial chemotaxis is composed of a core complex containing chemoreceptors, the histidine autokinase CheA, and the coupling protein CheW. Site-specific spin labeling and pulsed dipolar ESR spectroscopy (PDS) have been applied to investigate the structure of a soluble ternary complex formed by Thermotoga maritima CheA (TmCheA), CheW, and receptor signaling domains. Thirty-five symmetric spin-label sites (SLSs) were engineered into the five domains of the CheA dimer and CheW to provide distance restraints within the CheA:CheW complex in the absence and presence of a soluble receptor that inhibits kinase activity (Tm14). Additional PDS restraints among spin-labeled CheA, CheW, and an engineered single-chain receptor labeled at six different sites allow docking of the receptor structure relative to the CheA:CheW complex. Disulfide cross-linking between selectively incorporated Cys residues finds two pairs of positions that provide further constraints within the ternary complex: one involving Tm14 and CheW and another involving Tm14 and CheA. The derived structure of the ternary complex indicates a primary site of interaction between CheW and Tm14 that agrees well with previous biochemical and genetic data for transmembrane chemoreceptors. The PDS distance distributions are most consistent with only one CheW directly engaging one dimeric Tm14. The CheA dimerization domain (P3) aligns roughly antiparallel to the receptor-conserved signaling tip but does not interact strongly with it. The angle of the receptor axis with respect to P3 and the CheW-binding P5 domains is bound by two limits differing by ˜20°. In one limit, Tm14 aligns roughly along P3 and may interact to some extent with the hinge region near the P3 hairpin loop. In the other limit, Tm14 tilts to interact with the P5 domain of the opposite subunit in an interface that mimics that observed with the P5 homologue CheW. The time domain ESR data can be simulated from the model only if orientational variability is introduced for the P5 and, especially, P3 domains. The Tm14 tip also binds beside one of the CheA kinase domains (P4); however, in both bound and unbound states, P4 samples a broad range of distributions that are only minimally affected by Tm14 binding. The CheA P1 domains that contain the substrate histidine are also broadly distributed in space under all conditions. In the context of the hexagonal lattice formed by trimeric transmembrane chemoreceptors, the PDS structure is best accommodated with the P3 domain in the center of a honeycomb edge.
|
|
|
Scanned-Probe Detection of Electron Spin Resonance from a Nitroxide Spin Probe
E.W. Moore, S.G. Lee, S.A. Hickman, S.J. Wright, L.E. Harrell, P.P. Borbat, J.H. Freed, and J.A. Marohn.
PNAS 106, 22251-22256 (2009).
Supporting Information
<doi: 10.1073/pnas.0908120106>
PMID:
20018707
PMCID:
PMC2799694
|
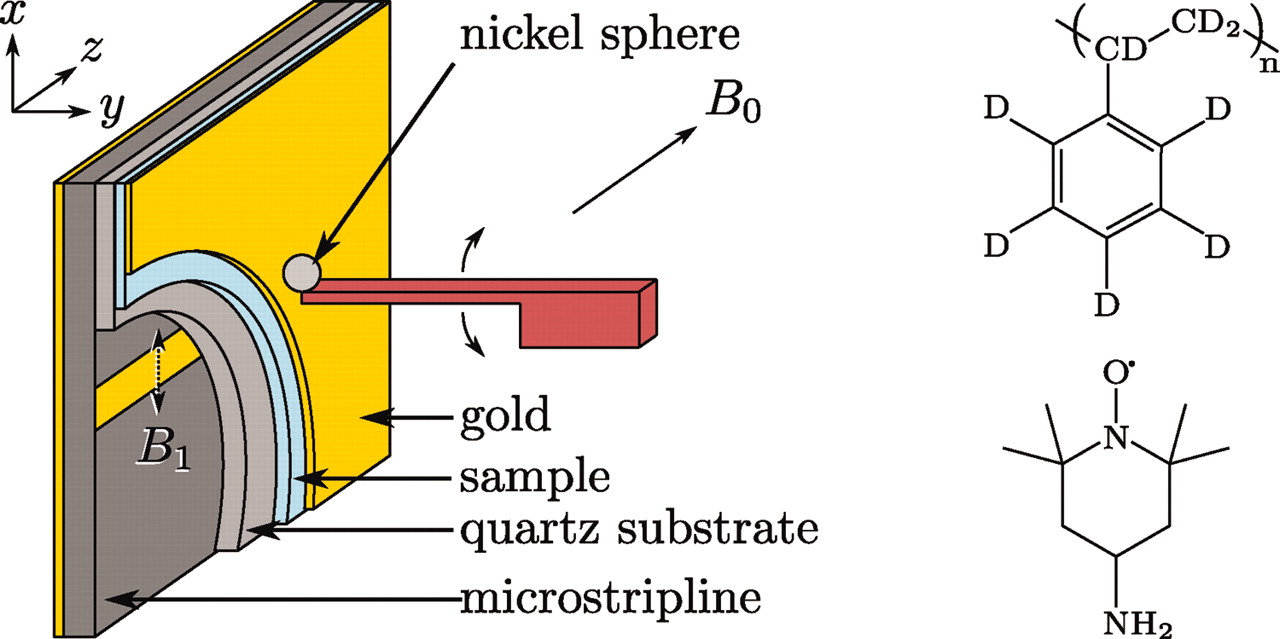
|
|
ABSTRACT: We report an approach that extends the applicability of ultrasensitive force-gradient detection of magnetic resonance to samples with spin-lattice relaxation times (T1) as short as a single cantilever period. To demonstrate the generality of the approach, which relies on detecting either cantilever frequency or phase, we used it to detect electron spin resonance from a T1 = 1 ms nitroxide spin probe in a thin film at 4.2 K and 0.6 T. By using a custom-fabricated cantilever with a 4 μm-diameter nickel tip, we achieve a magnetic resonance sensitivity of 400 Bohr magnetons in a 1 Hz bandwidth. A theory is presented that quantitatively predicts both the lineshape and the magnitude of the observed cantilever frequency shift as a function of field and cantilever-sample separation. Good agreement was found between nitroxide T1's measured mechanically and inductively, indicating that the cantilever magnet is not an appreciable source of spin-lattice relaxation here. We suggest that the new approach has a number of advantages that make it well suited to push magnetic resonance detection and imaging of nitroxide spin labels in an individual macromolecule to single-spin sensitivity.
|
|
|
Variable Coupling Scheme for High-Frequency Electron Spin Resonance Resonators Using Asymmetric Meshes
D.S. Tipikin, K.A. Earle and J.H. Freed.
Appl. Magn. Reson. 37, 819-832 (2010).
<doi: 10.1007/s00723-009-0088-1>
PMID:
20458356
PMCID:
PMC2865681
|
|
|
ABSTRACT: The sensitivity of a high-frequency electron spin resonance (ESR) spectrometer depends strongly on the structure used to couple the incident millimeter wave to the sample that generates the ESR signal. Subsequent coupling of the ESR signal to the detection arm of the spectrometer is also a crucial consideration for achieving high spectrometer sensitivity. In previous work, we found that a means for continuously varying the coupling was necessary for attaining high sensitivity reliably and reproducibly. We report here on a novel asymmetric mesh structure that achieves continuously variable coupling by rotating the mesh in its own plane about the millimeter-wave transmission-line optical axis. We quantify the performance of this device with nitroxide spin label spectra in both a lossy aqueous solution and a low-loss solid-state system. These two systems have very different coupling requirements and are representative of the range of coupling achievable with this technique. Lossy systems, in particular, are a demanding test of the achievable sensitivity and allow us to assess the suitability of this approach for applying high-frequency ESR, e.g., to the study of biological systems at physiological conditions. The variable coupling technique reported on here allows us to readily achieve a factor of ca. 7 improvement in the signal-to-noise ratio at 170 GHz and a factor of ca. 5 at 95 GHz over what has previously been reported for lossy samples.
|
|
|
Calculation of Double-Quantum-Coherence Two-dimensional Spectra: Distance Measurements and Orientational Correlations
S.K. Misra, P.P. Borbat, J.H. Freed.
Appl. Magn. Reson. 36, 237-258 (2009).
<doi: 10.1007/s00723-009-0023-5>
PMID:
20161423
PMCID:
PMC2786184
|

|
|
ABSTRACT: The double-quantum-coherence (DQC) echo signal for two coupled nitroxides separated by distances ≳10 Å, is calculated rigorously for the six-pulse sequence. Successive application of six pulses on the initial density matrix, with appropriate inter-pulse time evolution and coherence pathway selection leaves only the coherent pathways of interest. The amplitude of the echo signal following the last π pulse can be used to obtain a one-dimensional (1D) dipolar spectrum (Pake doublet), and the echo envelope can be used to construct the 2D DQC spectrum. The calculations are carried out using the product space spanned by the two electron-spin magnetic quantum numbers m1, m2 and the two nuclear-spin magnetic quantum numbers M1, M2, describing, e.g. two coupled nitroxides in bilabeled proteins. The density matrix is subjected to a cascade of unitary transformations taking into account dipolar and electron exchange interactions during each pulse and during the evolution in the absence of a pulse. The unitary transformations use the eigensystem of the effective spin Hamiltonians obtained by numerical matrix diagonalization. Simulations are carried out for a range of dipolar interactions, D, and microwave magnetic field strength B1 for both fixed and random orientations of the two 14N (and 15N) nitroxides. Relaxation effects were not included. Several examples of 1D and 2D Fourier transforms of the time-domain signals versus dipolar evolution and spin-echo envelope time variables are shown for illustration. Comparisons are made between 1D rigorous simulations and analytical approximations. The rigorous simulations presented here provide insights into DQC electron spin resonance spectroscopy, they serve as a standard to evaluate the results of approximate theories, and they can be employed to plan future DQC experiments.
|
|
|
A 236 GHz Fe3+ EPR Study of Nanoparticles of the Ferromagnetic Room-Temperature Semiconductor Sn1-xFexO2 (x = 0.005)
S.K. Misra, S.I. Andronenko, A. Punnoose, D. Tipikin, and J.H. Freed.
Appl. Magn. Reson. 36, 291-295 (2009).
<doi: 10.1007/s00723-009-0024-4>
PMID:
20161547
PMCID:
PMC2805097
|

|
|
ABSTRACT: High-frequency (236 GHz) electron paramagnetic resonance (EPR) studies of Fe3+ ions at 255 K are reported in a Sn1−xFexO2 powder with x = 0.005, which is a ferromagnetic semiconductor at room temperature. The observed EPR spectrum can be simulated reasonably well as the overlap of spectra due to four magnetically inequivalent high-spin (HS) Fe3+ ions (S = 5/2). The spectrum intensity is calculated, using the overlap I(BL) + (I(HS1) + I(HS2) + I(HS3) + I(HS4)) × exp(−0.00001B), where B is the magnetic field intensity in Gauss, I represents the intensity of an EPR line (HS1, HS2, HS3, HS4), and BL stands for the baseline (the exponential factor, as found by fitting to the experimental spectrum, is related to the Boltzmann population distribution of energy levels at 255 K, which is the temperature of the sample in the spectrometer). These high-frequency EPR results are significantly different from those at X-band. The large values of the zero-field splitting parameter (D) observed here for the four centers at the high frequency of 236 GHz are beyond the capability of X-band, which can only record spectra of ions with much smaller D values than those reported here.
|
|
|
Multifrequency ESR Study of Spin-Labeled Molecules in Inclusion Compounds with Cyclodextrins
B. Dzikovski, D. Tipikin, V. Livshits, K.A. Earle and J.H. Freed.
Phys. Chem. Chem. Phys. 11, 6676-6688 (2009).
Supporting Information
<doi: 10.1039/b903490k>
PMID:
19639141
PMCID:
PMC2743463
|

|
|
ABSTRACT: The molecular dynamics of spin-labeled compounds included into the solid phase of cyclodextrins (CDs) has been studied using conventional (X-band) ESR at 9 GHz and high-field high-frequency (HFHF) ESR at 240 and 170 GHz. The patterns of axial rotation at these higher frequencies are clear just by inspection of the spectrum, unlike the case for 9 GHz spectra. That is HFHF ESR is sensitive to molecular motion about the diffusion axis collinear with the X, Y or Z-direction of the magnetic g- and A-tensors of the nitroxide moiety (referred to, respectively, as X, Y or Z-rotation). For doxyl stearic acids (Z-rotation) and TEMPOyl caprylate (X-rotation) included in β- and γ-CDs we were able to determine the rate of molecular motion and the corresponding potential barriers. We emphasize that determining the rate of Z-rotation by ESR is feasible only using HFHF ESR. For the X-rotation case we suggest that the motion of the nitroxide moiety consists of fast small-angle librations about the magnetic X-axis superimposed by rotational diffusion about the same axis. The potential barrier of 1.7 Kcal mol−1 for this rotational diffusion is unusually low. A fascinating feature of TEMPO derivatives included in β-CD is the detectable molecular motion at temperatures below 77 K. For the other CD-spin probe systems, we used multifrequency analysis to assign the conformations of spin-labeled molecules. A dramatic spectral change for 16-sasl in β- and γ-CDs at ˜260 K corresponds to a tilting of the position of the nitroxide moiety on the rotating molecule relative to the long diffusion axis, while for TEMPO derivatives in γ-cyclodextrin below 200 K, we observe a rapid transition from fast to very slow rotational motion. More complex features are best studied by means of multifrequency ESR experiments. The visual clarity and the simplicity of analysis of the ESR spectra shown in this work should provide a benchmark for future studies of molecular motion by HFHF ESR.
|
|
|
Fusion Peptide from Influenza Hemagglutinin Increases Membrane Surface Order: An Electron-Spin Resonance Study
M. Ge and J.H. Freed.
Biophys. J. 96, 4925-4934 (2009).
Supporting Information
<doi: 10.1016/j.bpj.2009.04.015>
PMID:
19527651
PMCID:
PMC2712059
|

|
|
ABSTRACT: A spin-labeling study of interactions of a fusion peptide from the hemagglutinin of the influenza virus, wt20, and a fusion-inactive mutant ΔG1 with dimyristoylphosphatidylcholine (DMPC) and 1-palmitoyl-2-oleoyl-phosphatdylcholine bilayers was performed. We found that upon binding of wt20, the ordering of headgroups and the ordering of acyl chains near the headgroup increased significantly, in a manner consistent with a cooperative phenomenon. However, changes in the order at the end of the acyl chains were negligible. The ordering effect of wt20 on the headgroup was much stronger at pH 5 than at pH 7. No effect of ΔG1 binding on the order of bilayers was evident. We also found that 1-palmitoyl-2-hydroxyl phosphatidylcholine, a membrane-fusion inhibitor, decreased the ordering of DMPC headgroups, whereas arachidonic acid, a membrane-fusion promoter, increased the ordering of DMPC headgroups. These results suggest that increases in headgroup ordering may be important for membrane fusion. We propose that upon binding of wt20, which is known to affect only the outer leaflet of the bilayer, this outer leaflet becomes more ordered, and thus more solid-like. Then the coupling between the hardened outer leaflet and the softer inner leaflet generates bending stresses in the bilayer, which tend to increase the negative curvature of the bilayer. We suggest that the increased ordering in the headgroup region enhances dipolar interactions and lowers electrostatic energy, which may provide an energy source for membrane fusion. Possible roles of bending stresses in promoting membrane fusion are discussed.
|
|
|
Membrane Fluidity
B. Dzikovski and J.H. Freed.
In Wiley Encyclopedia of Chemical Biology; T. P. Begley and B. A. Baird, Eds.
Wiley; Hoboken, NJ, USA, 2009; Chapter 2, pp 728-741.
|
|
|
Membrane Fluidity
B. Dzikovski and J.H. Freed.
In Wiley Encyclopedia of Chemical Biology; T. P. Begley and B. A. Baird, Eds.
Wiley; Hoboken, NJ, USA, 2009; Chapter 2, pp 728-741.
<doi: 10.1002/9780470048672.wecb313>
PMID: [None - Book] PMCID:
[None - Book]
|
|
|
ABSTRACT: In the popular fluid mosaic model for biomembranes, membrane proteins and other membrane-embedded molecules are in a two-dimensional fluid formed by the phospholipids. Such a fluid state allows free motion of constituents within the membrane bilayer and is extremely important for membrane function. The term "membrane fluidity" is a general concept, which refers to the ease of motion for molecules in the highly anisotropic membrane environment. We give a brief description of physical parameters associated with membrane fluidity, such as rotational and translational diffusion rates, order parameters, etc., and review physical methods used for their determination. We also show limitations of the fluid mosaic model and discuss recent developments in membrane science that pertain to fluidity, such as evidence for compartmentalization of the biomembrane by the cell cytoskeleton.
|
|
|
A Scissors Mechanism for Stimulation of SNARE-Mediated Lipid Mixing by Cholesterol
J. Tong, P.P. Borbat, J.H. Freed, and Y.-K. Shin.
PNAS 106, 5141-5146 (2009).
Supporting Information
<doi: 10.1073/pnas.0813138106>
PMID:
19251653
PMCID:
PMC2663986
|
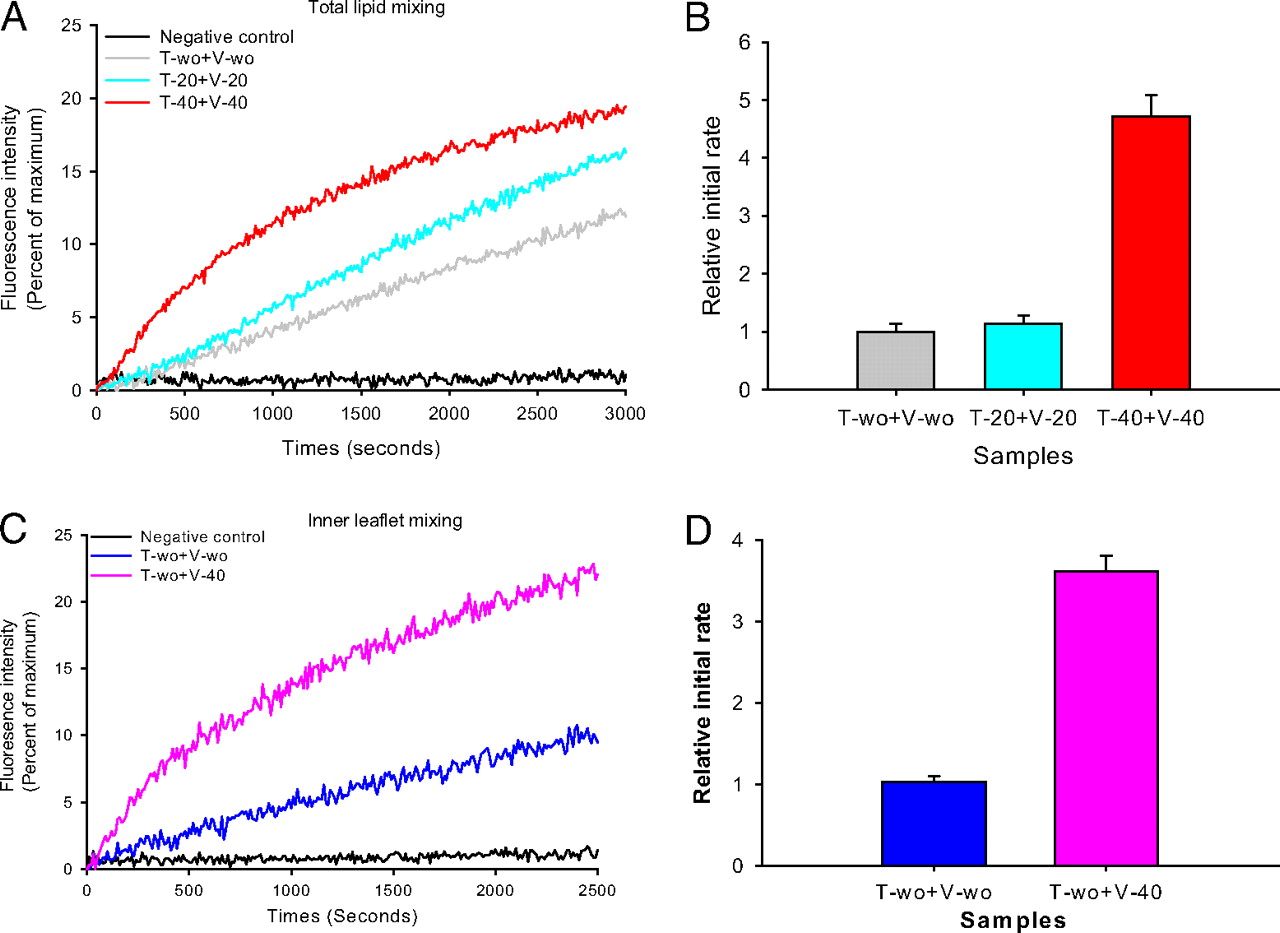
|
|
ABSTRACT: Neurotransmitter release at the synapse requires membrane fusion. The SNARE complex, composed of the plasma membrane t-SNAREs syntaxin 1A and SNAP-25 and the vesicle v-SNARE synaptobrevin, mediates the fusion of 2 membranes. Synaptic vesicles contain unusually high cholesterol, but the exact role of cholesterol in fusion is not known. In this study, cholesterol was found to stimulate SNARE-mediated lipid mixing of proteoliposomes by a factor of 5 at a physiological concentration. Surprisingly, however, the stimulatory effect was more pronounced when cholesterol was on the v-SNARE side than when it was on the t-SNARE side. Site-directed spin labeling and both continuous wave (CW) and pulsed EPR revealed that cholesterol induces a conformational change of the v-SNARE transmembrane domain (TMD) from an open scissors-like dimer to a parallel dimer. When the TMD was forced to form a parallel dimer by the disulfide bond, the rate was stimulated 2.3-fold even without cholesterol, supporting the relevance of the open-to-closed conformational change to the fusion activity. The open scissors-like conformation may be unfavorable for fusion and cholesterol may relieve this inhibitory factor.
|
|
|
Molecular-Scale Force Measurement in a Coiled-Coil Peptide Dimer by Electron Spin Resonance
S.V. Gullà, G. Sharma, P.Borbat, J.H. Freed, H. Ghimire, M.R. Benedikt, N.L. Holt, G.A. Lorigan, K. Rege, C. Mavroidis, and D.E. Budil.
J. Am. Chem. Soc. 131, 5374-5375 (2009).
|

|
|
Molecular-Scale Force Measurement in a Coiled-Coil Peptide Dimer by Electron Spin Resonance
S.V. Gullà, G. Sharma, P.Borbat, J.H. Freed, H. Ghimire, M.R. Benedikt, N.L. Holt, G.A. Lorigan, K. Rege, C. Mavroidis, and D.E. Budil.
J. Am. Chem. Soc. 131, 5374-5375 (2009).
Supporting Information
<doi: 10.1021/ja900230w>
PMID:
19331323
PMCID:
PMC2888838
|
|
|
ABSTRACT: A new method for measuring forces between small protein domains based on double electron−electron resonance (DEER) spectroscopy is demonstrated using a model peptide derived from the α-helical coiled-coil leucine zipper of yeast transcriptional activator GCN4. The equilibrium distribution of distances between two nitroxide spin labels rigidly attached to the helices of the dimer was determined by DEER and yielded a closing force of 100 ± 10 pN between monomers, in excellent agreement with theoretical predictions.
|
|
|
Multifrequency Electron Spin Resonance Spectra of a Spin-Labeled Protein Calculated from Molecular Dynamics Simulations
D. Sezer, J.H. Freed, and B. Roux.
J. Am. Chem. Soc. 131, 2597-2605 (2009).
Supporting Information
<doi: 10.1021/ja8073819>
PMID:
19191603
PMCID:
PMC2763593
|

|
|
ABSTRACT: Multifrequency electron spin resonance (ESR) spectra provide a wealth of structural and dynamic information about the local environment of the spin label and, indirectly, about the spin-labeled protein. Relating the features of the observed spectra to the underlying molecular motions and interactions is, however, challenging. To make progress toward a rigorous interpretation of ESR spectra, we perform extensive molecular dynamics (MD) simulations of fully solvated T4 Lysozyme, labeled with the spin label MTSSL at positions 72 and 131. These two sites have been the object of numerous experimental studies and are generally considered as prototypical solvent-exposed sites on the surfaces of α-helices. To extend the time window afforded by the MD simulations, stochastic Markov models reflecting the dynamics of the spin label side chains in terms of their rotameric states are constructed from the trajectories. The calculated multifrequency ESR spectra are in very good agreement with experiment for three different magnetic field strengths without adjusting any parameters. During the trajectories, the spin labels interconvert among a fairly large number of conformations and display a propensity to form interactions with protein residues other than their nearest neighbors along the helix. The detailed picture of the spin label emerging from the MD simulations provides useful insight into the molecular origins of the available spectroscopic and crystallographic data.
|
|
|
Determination of Tie-Line Fields for Coexisting Lipid Phases: An ESR Study
A.K. Smith and J.H. Freed.
J. Phys. Chem. B 113, 3957-3971 (2009).
<doi: 10.1021/jp808412x>
PMID:
19673072
PMCID:
PMC2772187
|

|
|
ABSTRACT: A novel method we refer to as the tie-line field (TLF) method has been developed to globally determine the tie lines of any three-component two-phase coexistence region by fitting electron-spin resonance (ESR) spectra obtained from compositions on the coexistence curve and within the coexistence region. The TLF method is illustrated by applying it to the liquid-ordered (Lo) and liquid-disordered (Ld) phase coexistence region of the lipid system brain-sphingomyelin / dioleoylphosphatidylcholine / cholesterol (SPM/DOPC/chol), for which an estimate of a tie-line was previously obtained by an earlier method also using ESR spectra. The essential aspect of the TLF method is the unique parametrization of the coexistence region called a "ruled surface". The use of the ruled surface enables one to guarantee that tie lines do not cross, as required by the phase rule, whereas previous methods lack this important constraint. It also makes simultaneous use of the full data set in determining the TLF and leads to a more efficient experimental design than previously used. The method is first tested out on synthetic data sets, then least-squares fitting of the ESR spectra with the parametrized model results in a tie-line field consistent with other known information on this lipid system. The best-fit tie-line field consists of the set of tie lines which are not exactly parallel; they exhibit a gradual change in slope with the largest slope within the coexistence region connecting the coexistence curve compositions with the highest and lowest cholesterol concentrations. The results are compared with those from more constrained methods of representing the tie-line fields as well as with the previous tie-line determination for the SPM/DOPC/chol system. An accurate determination of the tie-line field of phase coexistence regions in lipid systems is a necessary step in determining coexisting lipid compositions to serve as models of cell plasma membranes.
|
|
|
Membrane-Bound α-Synuclein Forms an Extended Helix: Long-Distance Pulsed ESR Measurements Using Vesicles, Bicelles, and Rodlike Micelles
E.R. Georgieva, T.F. Ramlall, P.P. Borbat, J.H. Freed, and D. Eliezer.
J. Am. Chem. Soc. 130, 12856-12857 (2008).
Supporting Information
<doi: 10.1021/ja804517m>
PMID:
18774805
PMCID:
PMC2626176
|
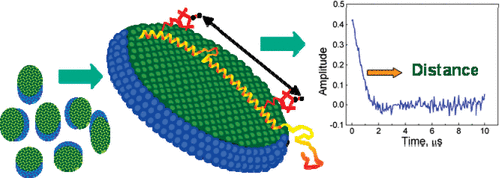
|
|
ABSTRACT: We apply pulsed dipolar ESR spectroscopy (Ku-band DEER) to elucidate the global conformation of the Parkinson's disease-associated protein, α-synuclein (αS) bound to small unilamellar phospholipid vesicles, rodlike SDS micelles, or lipid bicelles. By measuring distances as long as ˜7 nm between introduced pairs of nitroxide spin labels, we show that distances are close to the expectations for a single continuous helix in all cases studied. In particular, we find distances of 7.5 nm between sites 24 and 72; 5.5 nm between sites 24 and 61; and 2 nm between sites 35 and 50. We conclude that αS does not retain a "hairpin" structure with two antiparallel helices, as is known to occur with spheroidal micelles, in agreement with our earlier finding that the protein's geometry is determined by the surface topology rather than being constrained by the interhelix linker. While the possibility of local helix discontinuities in the structure of membrane-bound αS remains, our data are more consistent with one intact helix. Importantly, we demonstrate that bicelles produce very similar results to liposomes, while offering a major improvement in experimentally accessible distance range and resolution, and thus are an excellent lipid membrane mimetic for the purpose of pulse dipolar ESR spectroscopy.
|
|
|
Using Markov Models to Simulate Electron Spin Resonance Spectra from Molecular Dynamics Trajectories
D. Sezer, J.H. Freed, and B. Roux.
J. Phys. Chem. B 112, 11014-11027 (2008).
<doi: 10.1021/jp801608v>
PMID:
18698714
PMCID:
PMC2562300
|
|
|
ABSTRACT: Simulating electron spin resonance (ESR) spectra directly from molecular dynamics simulations of a spin-labeled protein necessitates a large number (hundreds or thousands) of relatively long (hundreds of nanoseconds) trajectories. To meet this challenge, we explore the possibility of constructing accurate stochastic models of the spin label dynamics from atomistic trajectories. A systematic, two-step procedure, based on the probabilistic framework of hidden Markov models, is developed to build a discrete-time Markov chain process that faithfully captures the internal spin label dynamics on time scales longer than about 150 ps. The constructed Markov model is used both to gain insight into the long-lived conformations of the spin label and to generate the stochastic trajectories required for the simulation of ESR spectra. The methodology is illustrated with an application to the case of a spin-labeled poly alanine α helix in explicit solvent.
|
|
|
Electron Spin Resonance Microscope for Imaging With Micron Resolution
A. Blank, C.R. Dunnam, P.P. Borbat, J.H. Freed.
Patent #7,403,008 B2, July 22, 2008
|

|
|
ABSTRACT: ESR microscope systems and methods for examining specimens using both continuous wave and pulsed modes in the 9 to 60 GHz range. The ESR microscope uses an image probe comprising gradient coils in addition to conventional modulation coils (in continuous wave mode) or magnetic field bias coils (in pulse mode), and a resonator constructed from high permittivity material. The systems and methods also involves the use of sample containers that permit the precise placement of samples in relation to the image probe. The microscope uses a microstrip or thin coaxial or dielectric antenna to obtain a high coupling coefficient to the specimen being imaged. The microscope systems provide resolution at the single micron level, and permit the observation of images comprising tens to hundreds of pixels for each of two or three dimensions in a few minutes. Novel stable radicals used as the imaging media are also described.
|
|
|
Simulating Electron Spin Resonance Spectra of Nitroxide Spin Labels from Molecular Dynamics and Stochastic Trajectories
D. Sezer, J.H. Freed, and B. Roux.
J. Chem. Phys. 128, 165106, (2008).
<doi: 10.1063/1.2908075>
PMID:
18447510
PMCID:
PMC2809671
|

|
|
ABSTRACT: Simulating electron spin resonance spectra of nitroxide spin labels from motional models is necessary for the quantitative analysis of experimental spectra. We present a framework for modeling the spin label dynamics by using trajectories such as those from molecular dynamics (MD) simulations combined with stochastic treatment of the global protein tumbling. This is achieved in the time domain after two efficient numerical integrators are developed: One for the quantal dynamics of the spins and the other for the classical rotational diffusion. For the quantal dynamics, we propagate the relevant part of the spin density matrix in Hilbert space. For the diffusional tumbling, we work with quaternions, which enables the treatment of anisotropic diffusion in a potential expanded as a sum of spherical harmonics. Time-averaging arguments are invoked to bridge the gap between the smaller time step of the MD trajectories and the larger time steps appropriate for the rotational diffusion and/or quantal spin dynamics.
|
|
|
Parametrization, Molecular Dynamics Simulation, and Calculation of Electron Spin Resonance Spectra of a Nitroxide Spin Label on a Polyalanine α-Helix
D. Sezer, J.H. Freed, B. Roux.
J. Phys. Chem. B 112, 5755-5767 (2008).
Supporting Information
<doi: 10.1021/jp711375x>
PMID:
18412413
PMCID:
PMC2766176
|

|
|
ABSTRACT: The nitroxide spin label 1-oxyl-2,2,5,5-tetramethylpyrroline-3-methyl-methanethiosulfonate (MTSSL), commonly used in site-directed spin labeling of proteins, is studied with molecular dynamics (MD) simulations. After developing force field parameters for the nitroxide moiety and the spin label linker, we simulate MTSSL attached to a polyalanine α-helix in explicit solvent to elucidate the factors affecting its conformational dynamics. Electron spin resonance spectra at 9 and 250 GHz are simulated in the time domain using the MD trajectories and including global rotational diffusion appropriate for the tumbling of T4 Lysozyme in solution. Analysis of the MD simulations reveals the presence of significant hydrophobic interactions of the spin label with the alanine side chains.
|
|
|
Determination of the Oligomeric States of Human and Rat Monoamine Oxidases in the Outer Mitochondrial Membrane and Octyl β-D-Glucopyranoside Micelles Using Pulsed Dipolar Electron Spin Resonance Spectroscopy
A.K. Upadhyay, P.P. Borbat, J. Wang, J.H. Freed, and D.E. Edmondson.
Biochemistry 47, 1554-1566 (2008).
<doi: 10.1021/bi7021377>
PMID:
18198902
PMCID:
[accepted before 04/08]
|

|
|
ABSTRACT: Human monoamine oxidase A (hMAOA) is considered to be unique among mammalian MAOs in having a non-conservative Glu-X-Lys mutation (X being 151 in MAOAs and 142 in MAOB's), which is suggested to be the reason for its monomeric structure. This hypothesis has been tested in this work. A pargyline based nitroxide spin labeled irreversible inhibitor (ParSL) was used as a MAO active site specific spin probe to measure intersubunit distances in detergent (octyl β-D-glucopyranoside, OGP) purified and OMM bound forms by a pulsed dipolar ESR spectroscopic (PDS) technique. In a parallel approach, the covalent flavin cofactor present in the MAO active sites was reduced to its respective anionic flavin semiquinone and used for measuring inter-flavin distances in detergent purified samples. The measured interspin distances are within 0.1–0.3 nm of those estimated from the available dimeric crystal structures of human MAOB and rat MAOA and show that all human and rat MAOs exist as dimers in the OMM. In the OGP micelle, however, human and rat MAOAs exist only partially (≤50%) as dimers, whereas human and rat MAOBs exist entirely as dimers. The Lys-151-Glu mutant of human MAOA and the Glu-142-Lys mutant of human MAOB exhibit similar spectral properties as the corresponding wild-type enzymes. Therefore, no role of the Glu-X residue in stabilizing dimeric structures of MAOs was found. The monomeric crystal structure reported for human MAOA is thus a result of its instability in the OGP micelles. The general applicability of the PDS technique to structural studies of membrane proteins in their native membrane environments and detergent purified forms is discussed.
|
|
|
Oxygen Effects on the EPR Signals from Wood Charcoals: Experimental Results and the Development of a Model
O.Y. Grinberg, B.B. Williams, A.E. Ruuge, S.A. Grinberg, D.E. Wilcox, H.M. Swartz, and J.H. Freed.
J. Phys. Chem. B 111, 13316-13324 (2007).
<doi: 10.1021/jp072656l>
PMID:
17973414
|
|
|
ABSTRACT: Charcoals prepared from certain tropical woods contain stable paramagnetic centers, and these have been characterized by EPR spectroscopy in the absence and presence of oxygen. The EPR-detectable spin density has been determined, as has been the temperature- and frequency-dependence of the oxygen broadening of the EPR signal, which is orders of magnitude larger than that observed with other materials, such as lithium phthalocyanine. Three Lorentzian components are required to fit the char EPR spectrum in the presence of oxygen, and the oxygen-dependence of the line width, intensity, and resonance position of the three components have been quantified. These results and the properties of porous carbonaceous materials are used to develop a model to explain the effect of oxygen on the char EPR spectral properties. The model is based on oxygen adsorption on the char surface according to a Langmuir isotherm and a dipolar interaction between the paramagnetic adsorbed gas and the charcoal spins. The three EPR components are correlated with the three known classes (sizes) of pores in charcoal, with the largest line broadening attributed to dipolar relaxation of spins in micropores, which have a larger specific surface area and a higher concentration of adsorbed oxygen. An attenuated, but similar, EPR response to oxygen by chars when they are immersed in aqueous solution is attributed to water competition with oxygen for adsorption on the char surface.
|
|
|
An Improved Picture of Methyl Dynamics in Proteins from Slowly Relaxing Local Structure Analysis of 2H Spin Relaxation
E. Meirovitch, Y.E. Shapiro, A. Polimeno, and J.H. Freed.
J. Phys. Chem. B 111, 12865-12875 (2007).
<doi: 10.1021/jp072156s>
PMID:
17941658
PMCID:
PMC2885794
|

|
|
ABSTRACT: Protein dynamics is intimately related to biological function. Core dynamics is usually studied with 2H spin relaxation of the 13CDH2 group, analyzed traditionally with the model-free (MF) approach. We showed recently that MF is oversimplified in several respects. This includes the assumption that the local motion of the dynamic probe and the global motion of the protein are decoupled, the local geometry is simple, and the local ordering is axially symmetric. Because of these simplifications MF has yielded a puzzling picture where the methyl rotation axis is moving rapidly with amplitudes ranging from nearly complete disorder to nearly complete order in tightly packed protein cores. Our conclusions emerged from applying to methyl dynamics in proteins the slowly relaxing local structure (SRLS) approach of Polimeno and Freed (Polimeno, A.; Freed, J. H. J. Phys. Chem. 1995, 99, 10995–11006.), which can be considered the generalization of MF, with all the simplifications mentioned above removed. The SRLS picture derived here for the B1 immunoglobulin binding domain of peptostreptococcal protein L, studied over the temperature range of 15–45 °C, is fundamentally different from the MF picture. Thus, methyl dynamics is characterized structurally by rhombic local potentials with varying symmetries and dynamically by tenfold slower rates of local motion. On average, potential rhombicity decreases, mode-coupling increases, and the rate of local motion increases with increasing temperature. The average activation energy for local motion is 2.0 ± 0.2 kcal/mol. Mode-coupling affects the analysis even at 15 °C. The accuracy of the results is improved by including in the experimental data set relaxation rates associated with rank 2 coherences.
|
|
|
2D-ELDOR Using Full Sc- Fitting and Absorption Lineshapes
Y.-W. Chiang, A. Costa-Filho, J.H. Freed.
J. Magn. Reson. 188, 231-245 (2007).
<doi: 10.1016/j.jmr.2007.06.014>
PMID:
17681478
|

|
|
ABSTRACT: Recent progress in developing 2D-ELDOR (2D electron–electron double resonance) techniques to better capture molecular dynamics in complex fluids, particularly in model and biological membranes, is reported. The new "full Sc− method", which corrects the spectral analysis for the phase distortion effects present in the experiments, is demonstrated to enhance the sensitivity of 2D-ELDOR in reporting on molecular dynamics in complex membrane environments. That is, instead of performing spectral fitting in the magnitude mode, our new method enables simultaneous fitting of both the real and imaginary components of the Sc− signal. The full Sc− fitting not only corrects the phase distortions in the experimental data but also more accurately determines instrumental dead times. The phase corrections applied to the Sc− spectrum enable the extraction of the pure absorption-mode spectrum, which is characterized by much better resolution than the magnitude-mode spectrum. In the absorption mode, the variation of homogeneous broadening, which reports on the dynamics of the spin probe, can even be observed by visual inspection. This new method is illustrated with results from model membranes of dipalmitoyl-sn-glycero-phosphatidylcholine (DPPC)–cholesterol binary mixtures, as well as with results from plasma membrane vesicles of mast cells. In addition to the dynamic parameters, which provide quantitative descriptions for membranes at the molecular level, the high-resolution absorption spectra themselves may be used as a "fingerprint" to characterize membrane phases and distinguish coexisting components in biomembranes. Thus we find that 2D-ELDOR is greatly improved with the new "full Sc− method" especially for exploring the complexity of model and biological membranes.
|
|
|
Conformational Motion of the ABC Transporter MsbA Induced by ATP Hydrolysis
P.P. Borbat, K. Surendhran, M. Bortolus, P. Zou, J.H. Freed, H.S. Mchaourab.
PLoS Biology 5, 2211-2219 (2007).
Supporting Information
<doi: 10.1371/journal.pbio.0050271>
PMID:
17927448
PMCID:
PMC2001213
|

|
|
ABSTRACT: We measured the amplitude of conformational motion in the ATP-binding cassette (ABC) transporter MsbA upon lipopolysaccharide (LPS) binding and following ATP turnover by pulse double electron-electron resonance and fluorescence homotransfer. The distance constraints from both methods reveal large-scale movement of opposite signs in the periplasmic and cytoplasmic part of the transporter upon ATP hydrolysis. LPS induces distinct structural changes that are inhibited by trapping of the transporter in an ATP post-hydrolysis intermediate. The formation of this intermediate involves a 33-Å distance change between the two ABCs, which is consistent with a dimerization-dissociation cycle during transport that leads to their substantial separation in the absence of nucleotides. Our results suggest that ATP-powered transport entails LPS sequestering into the open cytoplasmic chamber prior to its translocation by alternating access of the chamber, made possible by 10–20-Å conformational changes.
|
|
|
Two-Dimensional ELDOR in the Study of Model and Biological Membranes
Y.-W. Chiang, A.J. Costa-Filho, and J.H. Freed.
Appl. Magn. Reson. 31, 375-386 (2007).
<doi: 10.1007/BF03166591>
|

|
|
ABSTRACT: Recent studies on model and biological membranes by two-dimensional (2-D) electronelectron double resonance (ELDOR) are reviewed and discussed. The studies include (1) the phase behavior of dispersions of phospholipid-cholesterol membrane vesicles; (2) the effect of the ion-channel-forming peptide gramicidin A on the lipid membrane; and (3) the effects of stimulation by antigen of the immunoglobulin E receptors in plasma membrane vesicles upon the lipid structure. In the first studies it is shown that the 2-D ELDOR spectra enable clear distinctions amongst the different phases, and this leads to a reliable temperature and composition-dependent phase diagram. In the second studies it is possible to distinguish bulk and boundary lipids and to describe their different dynamic structures. In the third studies we could distinguish both liquid-ordered and liquid-disordered spectral components, and one finds that the fraction of the latter increases as a result of stimulation. Emphasis is placed on the new "full Sc — method" of processing the 2-D ELDOR data to significantly enhance spectral resolution, which is particularly important in studying the spectra from coexisting phases or components. In the full Sc — method, one utilizes both the real and imaginary parts of the signal, instead of their magnitude; the needed phase corrections are obtained as part of the nonlinear least-squares fitting of the 2-D ELDOR data.
|
|
|
Dynamic Molecular Structure and Phase Diagram of DPPC-Cholesterol Binary Mixtures: A 2D-ELDOR Study
Y.-W. Chiang, A.J. Costa-Filho, and J.H. Freed.
J. Phys. Chem. B 111, 11260-11270 (2007).
Supporting Information
<doi: 10.1021/jp0732110>
PMID:
17760438
|

|
|
ABSTRACT: This paper is an application of 2D electron−electron double resonance (2D-ELDOR) with the "full Sc− method" to study model membranes. We obtain and confirm the phase diagram of 1,2-dipalmitoyl-sn-glycerophosphatidylcholine (DPPC)−cholesterol binary mixtures versus temperature and provide quantitative descriptions for its dynamic molecular structure using 2D-ELDOR at the Ku band. The spectra from the end-chain 16-PC spin label in multilamellar phospholipid vesicles are obtained for cholesterol molar concentrations ranging from 0 to 50% and from 25 to 60 °C. This phase diagram consists of liquid-ordered, liquid-disordered, and gel phases and phase coexistence regions. The phase diagram is carefully examined according to the spectroscopic evidence, and the rigorous interpretation for the line shape changes. We show that the 2D-ELDOR spectra differ markedly with variation in the composition. The extensive line shape changes in the 2D-plus-mixing-time representation provide useful information to define and characterize the membrane phases with respect to their dynamic molecular structures and to determine the phase boundaries. The homogeneous T2's are extracted from the pure absorption spectra and are used to further distinguish the membrane phases. These results show 2D-ELDOR to be naturally suitable for probing and reporting the dynamic structures of microdomains in model membrane systems and, moreover, providing a very detailed picture of their molecular dynamic structure, especially with the aid of the "full Sc− method".
|
|
|
Rigid Body Refinement of Protein Complexes with Long-Range Distance Restraints from Pulsed Dipolar ESR
J. Bhatnagar, J.H. Freed, and B.R. Crane.
In Two-Component Signaling Systems, Part B, M. Simon, B.R. Crane and A.B. Crane, Eds.
Methods in Enzymology 423, Ch. 4, pp. 117-133 (2007).
<doi: 10.1016/S0076-6879(07)23004-6>
PMID:
17609128
|

|
|
ABSTRACT: The modeling of protein–protein complexes greatly benefits from the incorporation of experimental distance restraints. Pulsed dipolar electron spin resonance spectroscopy is one such powerful technique for obtaining long-distance restraints in protein complexes. Measurements of the dipolar interaction between two spins placed specifically within a protein complex give information about the spin–spin separation distance. We have developed a convenient method to incorporate such long-range distance information in the modeling of protein–protein complexes that is based on rigid body refinement of the protein components with the software Crystallography and NMR System (CNS). Factors affecting convergence such as number of restraints, error allocation scheme, and number and position of spin labeling sites were investigated with real and simulated data. The use of 4 to 5 different labeling sites on each protein component was found to provide sufficient coverage for producing accuracies limited by the uncertainty in the spin-label conformation within the complex. With an asymmetric scheme of allocating this uncertainty, addition of simulated restraints revealed the importance of longer distances within a limited set of total restraints. We present two case studies: (1) refinement of the complex formed between the histidine kinase CheA and its coupling protein CheW, and (2) refinement of intra-helical separations in the protein a-synuclein bound to micelles.
|
|
|
Measuring Distances by Pulsed Dipolar ESR Spectroscopy: Spin-Labeled Histidine Kinases
P.P. Borbat and J.H. Freed.
In Two-Component Signaling Systems, Part B, M. Simon, B.R. Crane and A.B. Crane, Eds.
Methods in Enzymology 423, Ch. 3, pp. 52-116 (2007).
<doi: 10.1016/S0076-6879(07)23003-4>
PMID:
17609127
|
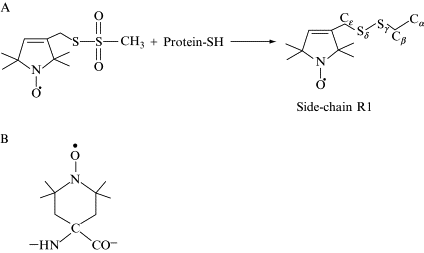
|
|
ABSTRACT: Applications of dipolar ESR spectroscopy to structural biology are rapidly expanding, and it has become a useful method that is aimed at resolving protein structure and functional mechanisms. The method of pulsed dipolar ESR spectroscopy (PDS) is outlined in the first half of the chapter, and it illustrates the simplicity and potential of this developing technology with applications to various biological systems. A more detailed description is presented of the implementation of PDS to reconstruct the ternary structure of a large dimeric protein complex from Thermotoga maritima, formed by the histidine kinase CheA and the coupling protein CheW. This protein complex is a building block of an extensive array composed of coupled supramolecular structures assembled from CheA/CheW proteins and transmembrane signaling chemoreceptors, which make up a sensor that is key to controlling the motility in bacterial chemotaxis. The reconstruction of the CheA/CheW complex has employed several techniques, including X-ray crystallography and pulsed ESR. Emphasis is on the role of PDS, which is part of a larger effort to reconstruct the entire signaling complex, including chemoreceptor, by means of PDS structural mapping. In order to precisely establish the mode of coupling of CheA to CheW and to globally map the complex, approximately 70 distances have already been determined and processed into molecular coordinates by readily available methods of distance geometry constraints.
|
|
|
Pros and Cons of Pulse Dipolar ESR: DQC and DEER
P.P. Borbat and J.H. Freed.
EPR Newsletter 17, 21-33 (2007).
|

|
|
INTRODUCTION: Continuous-wave (cw) and pulsed ESR have been extensively applied to biological problems in the context of molecular dynamics and are now increasingly applied to study biomolecular structure and function. cw ESR has been used to measure distances in the range of 6–20 Å between pairs of nitroxide spin labels. Distance measurements using pulse ESR methods, a major advance in this area, are currently able to deliver long-distance constraints in the range of 10–80 Å. The distance constraints from pulse ESR can for example be used to establish protein folding or orient and dock proteins and their subunits, yielding useful insights into the structure of a protein or a protein complex. They can also aid in refinement of NMR data. We refer to this emerging methodology as 'pulsed dipolar (ESR) spectroscopy' or PDS for short.
|
|
|
Modeling the Effects of Structure and Dynamics of the Nitroxide Side Chain on the ESR Spectra of Spin-Labeled Proteins
F. Tombolato, A. Ferrarini, and J.H. Freed.
J. Phys. Chem. B 110, 26260-26271 (2006).
<doi: 10.1021/jp064028u>
PMID:
17181284
PMCID:
PMC2885803
|
|
|
ABSTRACT: In the companion paper (J. Phys. Chem. B 2006, 110, jp0629487), a study of the conformational dynamics of methanethiosulfonate spin probes linked at a surface-exposed α-helix has been presented. Here, on the basis of this analysis, X-band ESR spectra of these spin labels are simulated within the framework of the Stochastic Liouville equation (SLE) methodology. Slow reorientations of the whole protein are superimposed on fast chain motions, which have been identified with conformational jumps and fluctuations in the minima of the chain torsional potential. Fast chain motions are introduced in the SLE for the protein reorientations through partially averaged magnetic tensors and relaxation times calculated according to the motional narrowing theory. The 72R1 and 72R2 mutants of T4 lysozyme, which bear the spin label at a solvent-exposed helix site, have been taken as test systems. For the side chain of the R2 spin label, only a few noninterconverting conformers are possible, whose mobility is limited to torsional fluctuations, yielding almost identical spectra, typical of slightly mobile nitroxides. In the case of R1, more complex spectra result from the simultaneous presence of constrained and mobile chain conformers, with relative weights that can depend on the local environment. The model provides an explanation for the experimentally observed dependence of the spectral line shapes on temperature, solvent, and pattern of substituents in the pyrroline ring. The relatively simple methodology presented here allows the introduction of realistic features of the spin probe dynamics into the simulation of ESR spectra of spin-labeled proteins; moreover, it provides suggestions for a proper account of such dynamics in more sophisticated approaches.
|
|
|
Dynamics of the Nitroxide Side Chain in Spin-Labeled Proteins
F. Tombolato, A. Ferrarini, and J.H. Freed.
J. Phys. Chem. B 110, 26248-26259 (2006).
<doi: 10.1021/jp0629487>
PMID:
17181283
PMCID:
PMC2883179
|
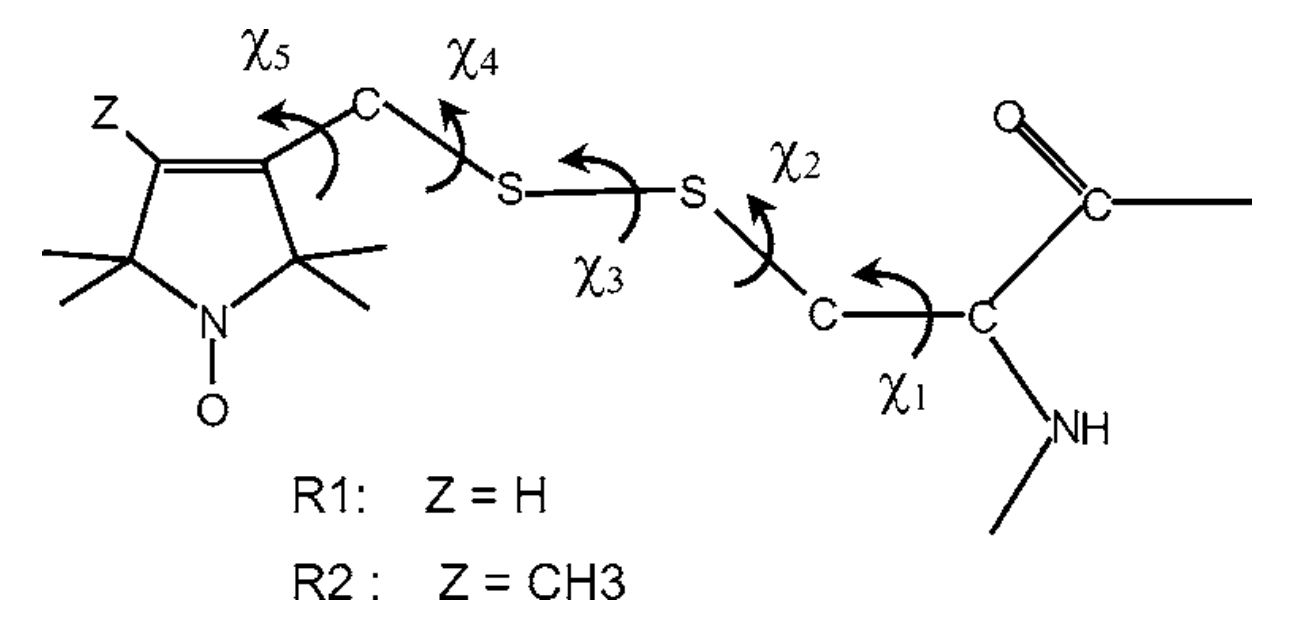
|
|
ABSTRACT: The dynamics of the tether linking methanethiosulfonate (MTSSL) spin probes to α-helices has been investigated with the purpose of rationalizing its effects on ESR line shapes. Torsional profiles for the chain bonds have been calculated ab initio, and steric interactions with the α-helix and the neighboring residues have been introduced at the excluded-volume level. As a consequence of the restrictions deriving from chain geometry and local constraints, a limited number of allowed conformers has been identified that undergo torsional oscillations and conformational jumps. Torsional fluctuations are described as damped oscillations, while transition rates between conformers are calculated according to the Langer multidimensional extension of the Kramers theory. The time scale and amplitude of the different motions are compared; the major role played by rotations of the outermost bonds of the side chain emerges, along with the effects of substituents in the pyrroline ring on the conformer distribution and dynamics. The extent and symmetry of magnetic tensor averaging produced by the side chain motions are estimated, the implications for the ESR spectra of spin-labeled proteins are discussed, and suggestions for the introduction of realistic features of the spin probe dynamics into the line shape simulation are presented.
|
|
|
|
|
Pulse Electron Spin Resonance Method for Investigation of Atoms in Impurity-Helium Solids
E.P. Bernard, V.V. Khmelenko, E. Vehmanen, P.P. Borbat, J.H. Freed, and D.M. Lee.
Proc. 24th Intl. Conf. on Low Temp. Physics, 1659-1660 (2006).
<doi: 10.1063/1.2355345>
|
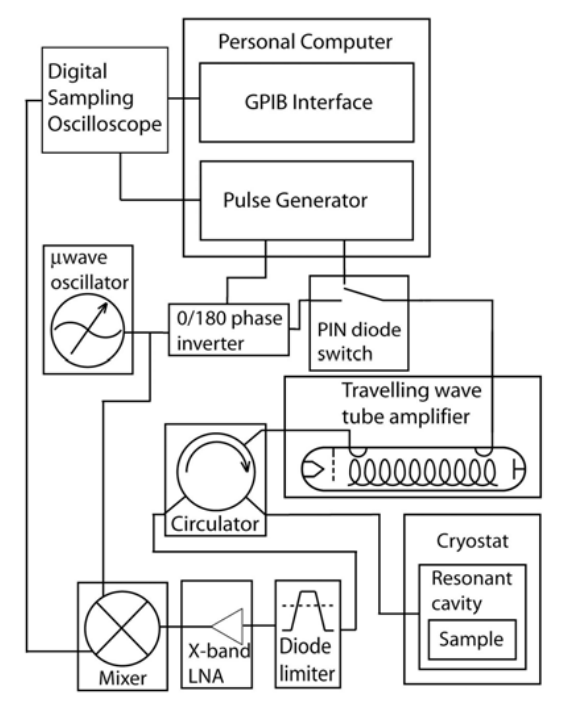
|
|
ABSTRACT: We have constructed an X-band pulse electron spin resonance spectrometer for the investigation of hydrogen, deuterium, and nitrogen impurity-helium solids at low temperatures. The spectrometer is of a conventional homodyne detection design incorporating a sampling oscilloscope but uses a modified CW ESR resonant cavity.
|
|
|
Collective Fluctuations in Ordered Fluids Investigated by Two-Dimensional Electron-Electron Double Resonance Spectroscopy
B. Fresch, D. Frezzato, G.J. Moro, G. Kothe, and J.H. Freed.
J. Phys. Chem. B 110, 24238-24254, (2006).
<doi: 10.1021/jp064028u>
PMID:
17125397
|
|
|
ABSTRACT: Two-dimensional electron−electron double resonance (2D-ELDOR) is a technique that is sensitive to the dynamical processes affecting spin labels in complex fluid environments. In ordered fluids, such as membrane vesicles, the 2D-ELDOR experiment is affected by the molecular tumbling in the locally ordered environment. This motion occurs on two different time scales, the faster molecular motion relative to the local director, and the slower collective fluctuations of the director field. In the experimental study of Patyal, Crepeau, and Freed (Biophys. J. 1997, 73, 2201), it was found that the widths of the autopeaks of the 2D-ELDOR spectrum increased as a function of the mixing time. In the present work, a theory is developed for the effects of director fluctuations on the autopeaks in the 2D-ELDOR experiment by employing an analytical solution of the stochastic Liouville equation for which the director field is treated as a multidimensional Gaussian process, as previously developed by Frezzato, Kothe, and Moro (J. Phys. Chem. B 2001, 105, 1281 and J. Phys. Chem. B 2004, 108, 9505). Good agreement is found between theory and experiment, notably the only adjustable parameter is κ, the bending elastic modulus of the membrane. The values of κ = 11 × 10-20 J for 1,2-dipalmitoyl-sn-glycero-phosphatidylcholine (DPPC) vesicles and κ = 15 × 10-20 J for DPPC/gramicidin A (5:1) vesicles, both at 45 °C, were found from the analysis and agree well with previous related measurements by other physical techniques. This establishes 2D-ELDOR as a useful technique to study the elastic properties of biological membranes.
|
|
|
Methyl Dynamics in Proteins from NMR Slowly Relaxing Local Structure Spin Relaxation Analysis: A New Perspective
Eva Meirovitch, Antonino Polimeno, and Jack H. Freed.
J. Phys. Chem. B 110, 20615-20628 (2006).
<doi: 10.1021/jp061403+>
PMID:
17034251
|
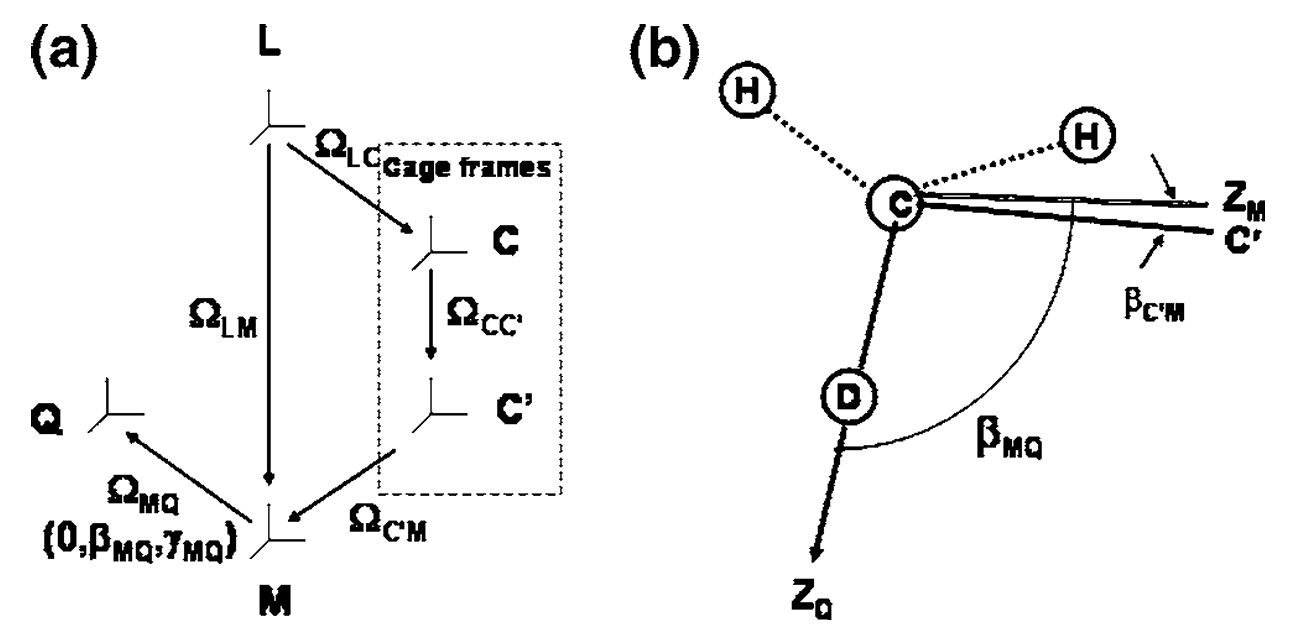
|
|
ABSTRACT: NMR spin relaxation of 2H nuclei in 13CH2D groups is a powerful method for studying side-chain motion in proteins. The analysis is typically carried out with the original model-free (MF) approach adapted to methyl dynamics. The latter is described in terms of axial local motions around, and of, the methyl averaging axis, mutually decoupled and independent of the global motion of the protein. Methyl motion is characterized primarily by the axial squared order parameter, S2axis, associated with fluctuations of the methyl averaging axis. This view is shown to be oversimplified by applying to typical experimental data the slowly relaxing local structure (SRLS) approach of Polimeno and Freed (Adv. Chem. Phys. 1993, 83, 89) which can be considered the generalization of the MF approach. Neglecting mode coupling and the asymmetry of the local ordering and treating approximately features of local geometry imply inaccurate values of S2axis, hence of the residual configurational entropy derived from it. S2axis, interpreted as amplitude of motion, was found to range from near disorder to almost complete order. Contrary to this picture, we find with the SRLS approach a moderate distribution in the magnitude of asymmetric local ordering and significant variation in its symmetry. The latter important property can be associated implicitly with the contribution of side-chain rotamer jumps. This is consistent with experimental residual dipolar coupling studies and theoretical work based on molecular dynamics simulations and molecular mechanics considerations. Configurational entropy is obtained in the SRLS approach directly from experimentally determined asymmetric potentials. Inconsistency between order parameters from 2H relaxation and from ηHC-HH cross-correlation and increase in order parameters with increasing temperature were observed with the MF approach. These discrepancies are reconciled, and physically tenable temperature dependence is obtained with the SRLS approach.
|
|
|
Two-Pulse Electron Spin Echo Study of Deuterium-Helium Solids
E.P. Bernard, V.V. Khmelenko, E. Vehmanen, P.P. Borbat, J.H. Freed, and D.M. Lee.
Proc. 24th Intl. Conf. on Low Temp. Physics, 372-373 (2006).
<doi: 10.1063/1.2354742>
|
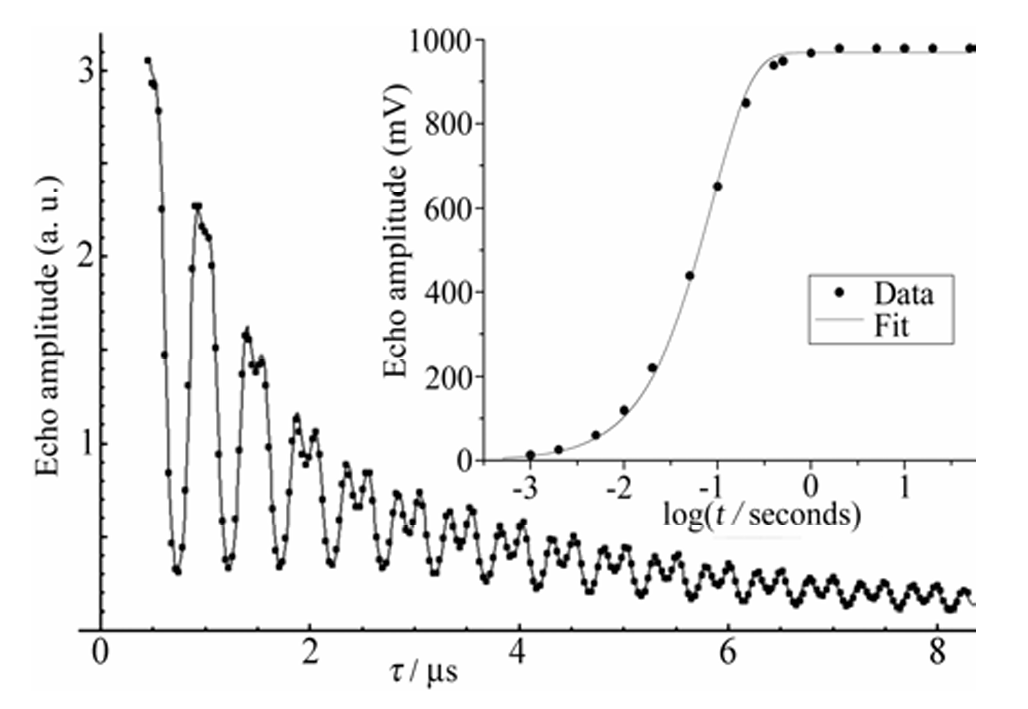
|
|
ABSTRACT: We measured electron spin echoes from deuterium atoms within deuterium-helium solids with an X-band pulse ESR spectrometer. Our two-pulse electron spin echo envelope modulation (ESEEM) measurements are well described by a model that places 50% – 60% of the deuterium atoms at the interface between the molecular deuterium nanoclusters and the superfluid liquid helium. The remainder of the atoms lie in substitutional sites within the nanoclusters. We also report the spin-lattice relaxation times T1 for these atoms.
|
|
|
Inter-Helix Distances in Lysophospholipid Micelle-Bound α-Synuclein from Pulsed ESR Measurements
P. Borbat, T.F. Ramlall, J.H. Freed, and D. Eliezer.
J. Am. Chem. Soc. 128, 10004-05 (2006).
Supporting Information
<doi: 10.1021/ja063122l>
PMID:
16881616
|
|
|
ABSTRACT: We demonstrate the use of pulsed ESR spectroscopy to measure intramolecular distances in the Parkinson's disease-associated protein α-synuclein bound to detergent and lysophospholipid micelles. We show that the inter-helical separation between the two helices formed upon binding to micelles is dependent on micelle composition, with micelles formed from longer acyl chains leading to an increased splaying of the two helices. Our data suggest that the topology of α-synuclein is not strongly constrained by the linker region between the two helices and instead depends on the geometry of the surface to which the protein is bound.
|
|
|
Coexisting Domains in the Plasma Membranes of Live Cells Characterized by ESR Spectroscopy
M.J. Swamy, L. Ciani, M. Ge, A.K. Smith, D. Holowka, B. Baird, and J.H. Freed.
Biophys. J. 90, 4452-4465 (2006).
Supporting Information
<doi: 10.1529/biophysj.105.070839>
PMID:
16565045
PMCID:
PMC1471862
|
|
|
ABSTRACT: The importance of membrane-based compartmentalization in eukaryotic cell function has become broadly appreciated, and a number of studies indicate that these eukaryotic cell membranes contain coexisting liquid-ordered (Lo) and liquid-disordered (Ld) lipid domains. However, the current evidence for such phase separation is indirect, and so far there has been no direct demonstration of differences in the ordering and dynamics for the lipids in these two types of regions or their relative amounts in the plasma membranes of live cells. In this study, we provide direct evidence for the presence of two different types of lipid populations in the plasma membranes of live cells from four different cell lines by electron spin resonance. Analysis of the electron spin resonance spectra recorded over a range of temperatures, from 5 to 37°C, shows that the spin-labeled phospholipids incorporated experience two types of environments, Lo and Ld, with distinct order parameters and rotational diffusion coefficients but with some differences among the four cell lines. These results suggest that coexistence of lipid domains that differ significantly in their dynamic order in the plasma membrane is a general phenomenon. The Lo region is found to be a major component in contrast to a model in which small liquid-ordered lipid rafts exist in a 'sea' of disordered lipids. The results on ordering and dynamics for the live cells are also compared with those from model membranes exhibiting coexisting Lo and Ld phases.
|
|
|
Protein Dynamics from NMR: The Slowly Relaxing Local Structure Analysis Compared with Model-Free Analysis
E. Meirovitch, Y.E. Shapiro, A. Polimeno, and J.H. Freed.
J. Phys. Chem. A 110, 8366-8396 (2006).
<doi: 10.1021/jp056975t>
PMID:
16821820
PMCID:
PMC2758167
|
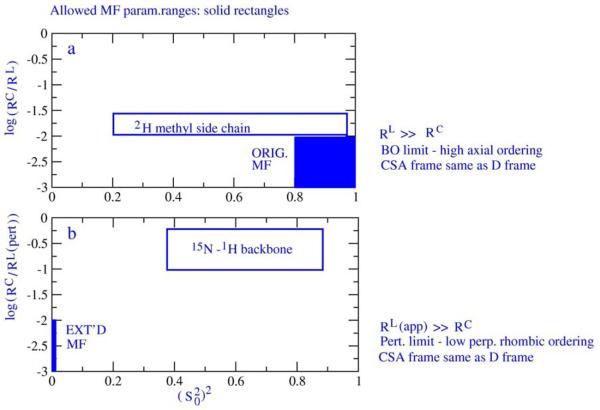
|
|
ABSTRACT: 15N−1H spin relaxation is a powerful method for deriving information on protein dynamics. The traditional method of data analysis is model-free (MF), where the global and local N−H motions are independent and the local geometry is simplified. The common MF analysis consists of fitting single-field data. The results are typically field-dependent, and multifield data cannot be fit with standard fitting schemes. Cases where known functional dynamics has not been detected by MF were identified by us and others. Recently we applied to spin relaxation in proteins the slowly relaxing local structure (SRLS) approach, which accounts rigorously for mode mixing and general features of local geometry. SRLS was shown to yield MF in appropriate asymptotic limits. We found that the experimental spectral density corresponds quite well to the SRLS spectral density. The MF formulas are often used outside of their validity ranges, allowing small data sets to be force-fitted with good statistics but inaccurate best-fit parameters. This paper focuses on the mechanism of force-fitting and its implications. It is shown that MF analysis force-fits the experimental data because mode mixing, the rhombic symmetry of the local ordering and general features of local geometry are not accounted for. Combined multifield multitemperature data analyzed with the MF approach may lead to the detection of incorrect phenomena, and conformational entropy derived from MF order parameters may be highly inaccurate. On the other hand, fitting to more appropriate models can yield consistent physically insightful information. This requires that the complexity of the theoretical spectral densities matches the integrity of the experimental data. As shown herein, the SRLS spectral densities comply with this requirement.
|
|
|
Reconstruction of the Chemotaxis Receptor-Kinase Assembly
S.-Y. Park, P.P. Borbat, G. Gonzalez-Bonet, J. Bhatnagar, A.M. Pollard, J.H. Freed, A.M. Bilwes & B.R. Crane.
Nat. Struct. Molec. Biol. 13, 400-407 (2006).
Supporting Information
<doi: 10.1038/nsmb1085>
PMID:
16622408
|
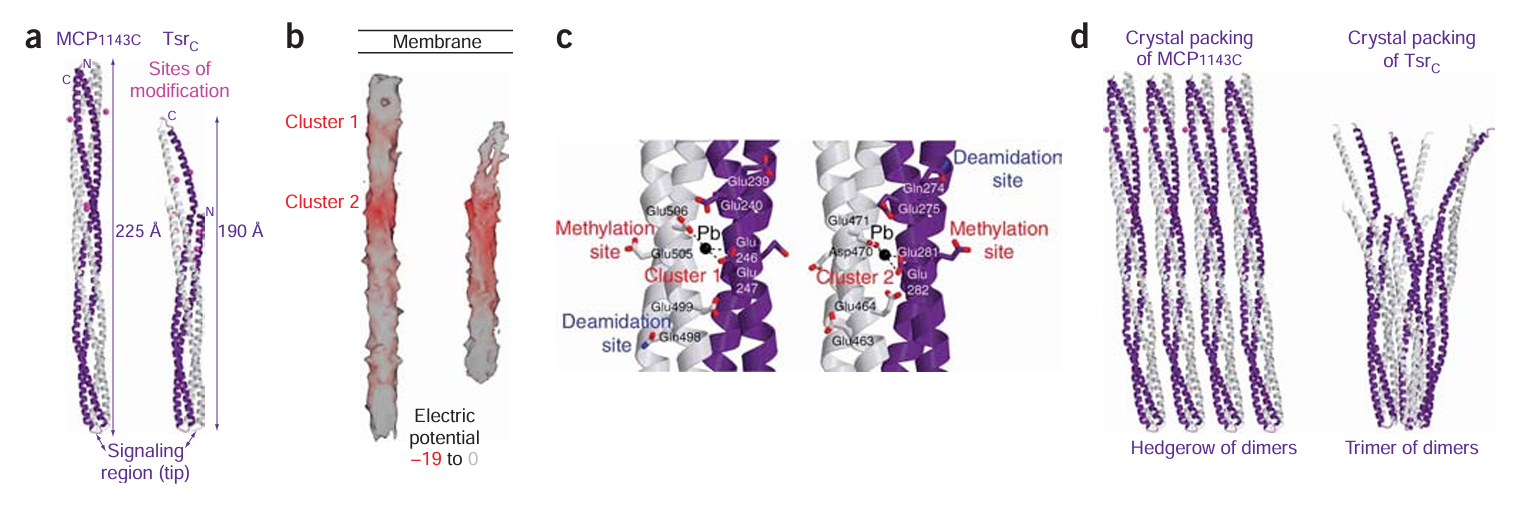
|
|
ABSTRACT: In bacterial chemotaxis, an assembly of transmembrane receptors, the CheA histidine kinase and the adaptor protein CheW processes environmental stimuli to regulate motility. The structure of a Thermotoga maritima receptor cytoplasmic domain defines CheA interaction regions and metal ion–coordinating charge centers that undergo chemical modification to tune receptor response. Dimeric CheA–CheW, defined by crystallography and pulsed ESR, positions two CheWs to form a cleft that is lined with residues important for receptor interactions and sized to clamp one receptor dimer. CheW residues involved in kinase activation map to interfaces that orient the CheW clamps. CheA regulatory domains associate in crystals through conserved hydrophobic surfaces. Such CheA self-contacts align the CheW receptor clamps for binding receptor tips. Linking layers of ternary complexes with close-packed receptors generates a lattice with reasonable component ratios, cooperative interactions among receptors and accessible sites for modification enzymes.
|
|
|
High-Field ESR on Aligned Membranes: A Simple Method to Record Spectra from Different Membrane Orientations in the Magnetic Field
B. Dzikovski, K. Earle, S. Pachtchenko, J. Freed.
J. Magn. Reson. 179, 273-279 (2006).
<doi: 10.1016/j.jmr.2005.12.015>
PMID:
16427793
|
|
|
ABSTRACT: A combination of isopotential spin-dry ultracentrifugation (ISDU) and microtome techniques was used to facilitate the collection of high field/high frequency (170 GHz) ESR spectra corresponding to different orientations of the membrane normal relative to the magnetic field. This technique is particularly valuable for aligned biological samples in vitro. At 170 GHz, conventional sample preparation techniques based solely on ISDU constrained the sample to be oriented so that the membrane normal was parallel to the applied magnetic field due to the geometry and the millimeter wave field distribution of the Fabry–Pérot resonator used in these experiments. This orientational constraint limited the information that could be obtained from aligned membranes at high field. The combined ISDU/microtome technique overcame this limitation. Spectra from spin-labeled Gramicidin A and the spin label cholestane in aligned DPPC membranes provide a demonstration of the technique. We also discuss some virtues of high field/high frequency ESR on aligned membranes compared to X-band.
|
|
|
Electron Spin Resonance Microscopy Applied to the Study of Controlled Drug Release
A. Blank, J.H. Freed, N.P. Kumar, C.-H. Wang.
J. Cont. Rel. 111, 174-184 (2006).
<doi: 10.1016/j.jconrel.2005.11.019>
PMID:
16460828
|
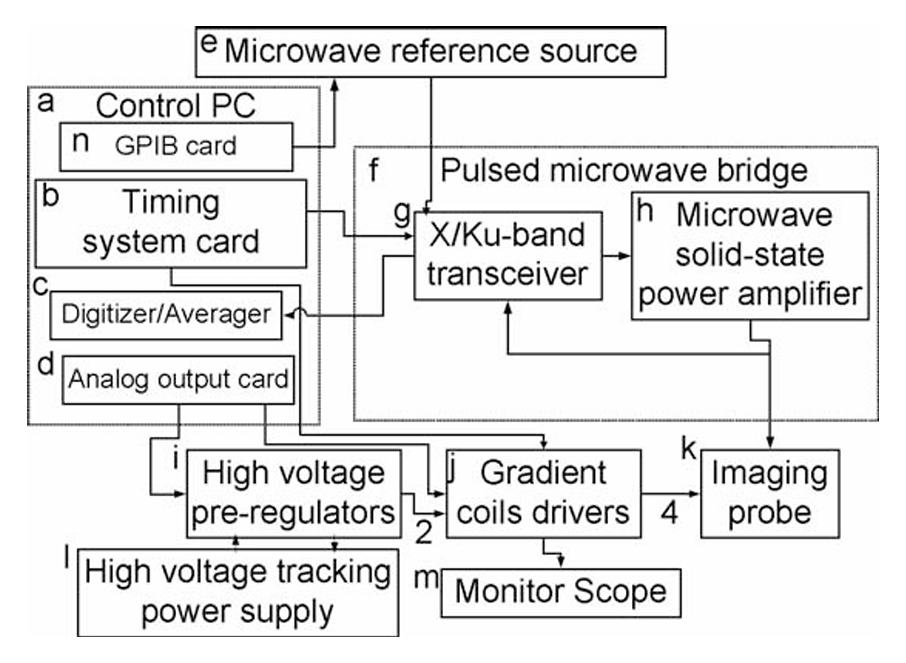
|
|
ABSTRACT: We describe our recent developments towards 3D micron-scale imaging capability, based on electron spin resonance (ESR), and its application to the study of controlled release. The method, termed ESR microscopy (ESRM), is an extension of the conventional "millimeter-scale" ESR imaging technique. It employs paramagnetic molecules (such as stable radicals or spin-labeled drugs) and may enable one to obtain accurate 3D spatially resolved information about the drug concentration, its self-diffusion tensor, rotational correlation time and the pH in the release matrix. Theoretical calculations, along with initial experimental results, suggest that a 3D resolution of ˜ 1 μm is feasible with this method. Here we were able to image successfully a high spin concentration sample with a resolution of ˜ 3 × 3 × 8 μm and subsequently study a single ˜ 120 μm biodegradable microsphere, internalized with a dilute solution of trityl radical, with a resolution of ˜ 12.7 × 13.2 × 26 μm. Analysis of the microsphere ESR imaging data revealed a likely increase in the viscosity inside the sphere and/or binding of the radical molecule to the sphere matrix. Future directions for progress are also discussed.
|
|
|
ESR Microscopy and Nanoscopy with "Induction" Detection
A. Blank and J.H. Freed.
Isr. J. Chem. 46, 423-438 (2006).
<doi: 10.1560/IJC_46_4_423>
|

|
|
ABSTRACT: The current state-of-the-art in the fields of Nuclear Magnetic Resonance (NMR) and Electron Spin Resonance (ESR) micro-imaging is reviewed. Special attention is given to the uniqueness and the advantages of the conventional "induction" detection method with respect to other emerging sensitive magnetic resonance detection and imaging techniques. Following this, a theoretical description of the factors affecting the sensitivity and resolution in induction detection ESR is provided. Based on the theory, new approaches to substantially improve ESR capabilities, both at room temperature and at low cryogenic temperatures, are discussed. Representative results of experiments conducted at room temperature and at frequencies of 10–16 GHz show that with test samples, a sensitivity of ˜107 electron spins and a resolution of ˜3 microns are currently available. The results confirm the validity of the theoretical approach and confirm the value of striving for even higher frequencies and lower temperatures, in order to further improve the performance Finally, some of the current and potential applications of ESR microscopy and nanoscopy (involving imaging with a resolution of ˜100 nm or better) are presented.
|
|
|
Maximum Entropy: A Complement to Tikhonov Regularization for Determination of Pair Distance Distributions by Pulsed ESR
Y.-W. Chiang, P.P. Borbat, J.H. Freed.
J. Magn. Reson. 177, 184-196 (2005).
<doi: 10.1016/j.jmr.2005.07.021>
PMID:
16137901
|
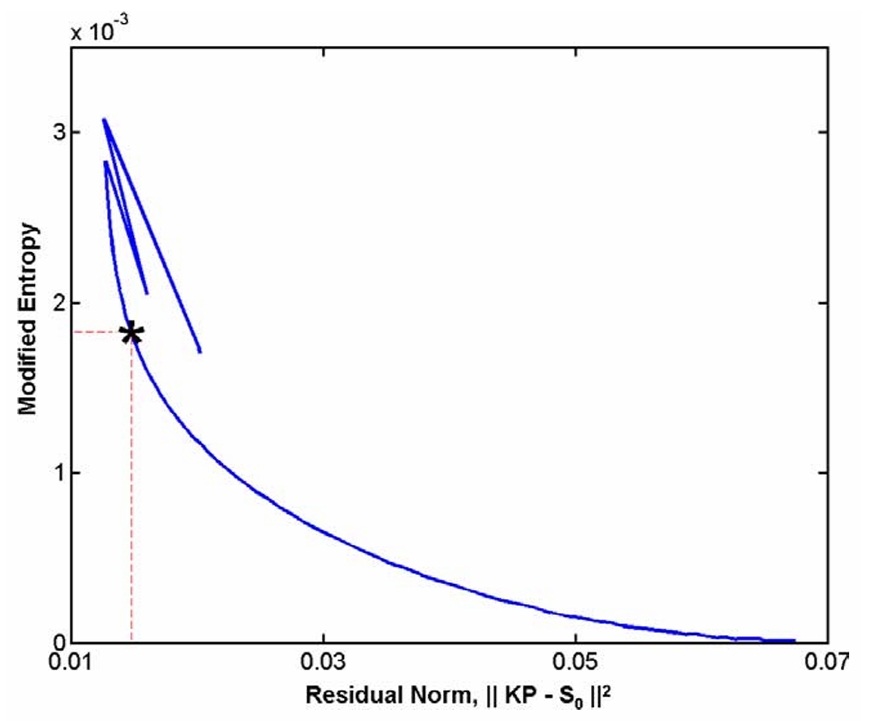
|
|
ABSTRACT: Tikhonov regularization (TIKR) has been demonstrated as a powerful and valuable method for the determination of distance distributions of spin-pairs in bi-labeled biomolecules directly from pulsed ESR signals. TIKR is a direct method, which requires no iteration, and, therefore, provides a rapid and unique solution. However, the distribution obtained tends to exhibit oscillatory excursions with negative portions in the presence of finite noise, especially in the peripheral regions of the distribution. The Shannon–Jaynes entropy of a probability distribution provides an intrinsic non-negativity constraint on the probability distribution and an unbiased way of obtaining information from incomplete data. We describe how the maximum entropy regularization method (MEM) may be applied to solve the ill-posed nature of the dipolar signal in pulsed ESR. We make use of it to suppress the negative excursions of the distance distribution and to increase the tolerance to noise in the dipolar signal. Model studies and experimental data are investigated, and they show that, with the initial or "seed" probability distribution that is required for MEM taken as the TIKR result, then MEM is able to provide a regularized solution, subject to the non-negativity constraint, and it is effective in dealing with noise that is problematic for TIKR. In addition we have incorporated into our MEM method the ability to extract the intermolecular dipolar component, which is embedded in the raw experimental data. We find that MEM minimization, which is implemented iteratively, is greatly accelerated using the TIKR result as the seed, and it converges more successfully. Thus we regard the MEM method as a complement to TIKR by securing a positive pair distance distribution and enhancing the accuracy of TIKR.
|
|
|
High-Frequency ESR at ACERT
K.A. Earle, B. Dzikovski, W. Hofbauer, J.K. Moscicki and J.H. Freed.
Magn. Reson. Chem. 43, S256-S266 (2005).
|
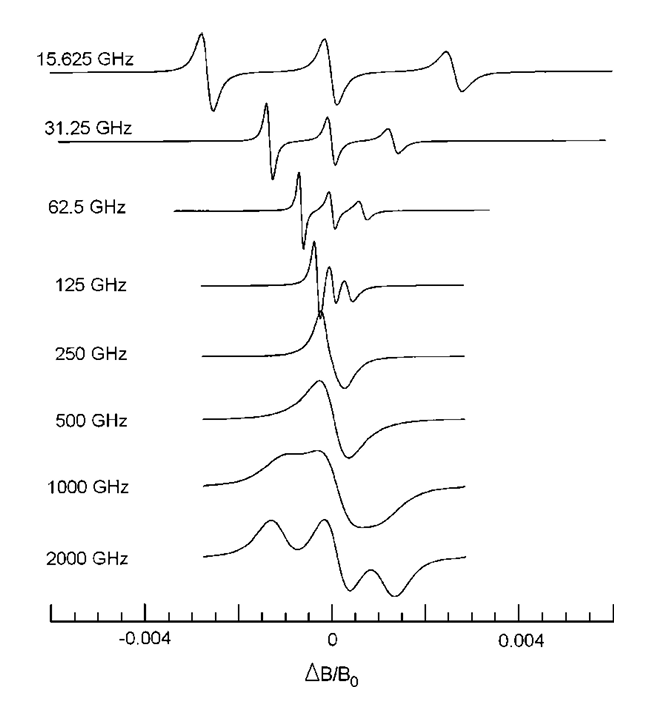
|
|
High-Frequency ESR at ACERT
K.A. Earle, B. Dzikovski, W. Hofbauer, J.K. Moscicki and J.H. Freed.
Magn. Reson. Chem. 43, S256-S266 (2005).
<doi: 10.1002/mrc.1684>
PMID:
16235203
|
|
|
ABSTRACT: High-field ESR offers many advantages in exploring fundamental questions of structure and dynamics in chemical, biological and physical samples. We provide a review of recent work performed at ACERT demonstrating the utility and flexibility of our methods for extracting both qualitative and quantitative information from a variety of systems. In particular, we emphasize the utility of multi-frequency ESR techniques for unraveling the details of the complex dynamical modes of proteins in solution and in heterogeneous systems such as lipid bilayers. We also include indications of directions for future work where appropriate.
|
|
|
Effects of Finite Pulse Width on Two-Dimensional Fourier Transform Electron Spin Resonance
Z. Liang, R.H. Crepeau, J.H. Freed.
J. Magn. Reson. 177, 247-260 (2005).
<doi: 10.1016/j.jmr.2005.07.024>
PMID:
16150620
|
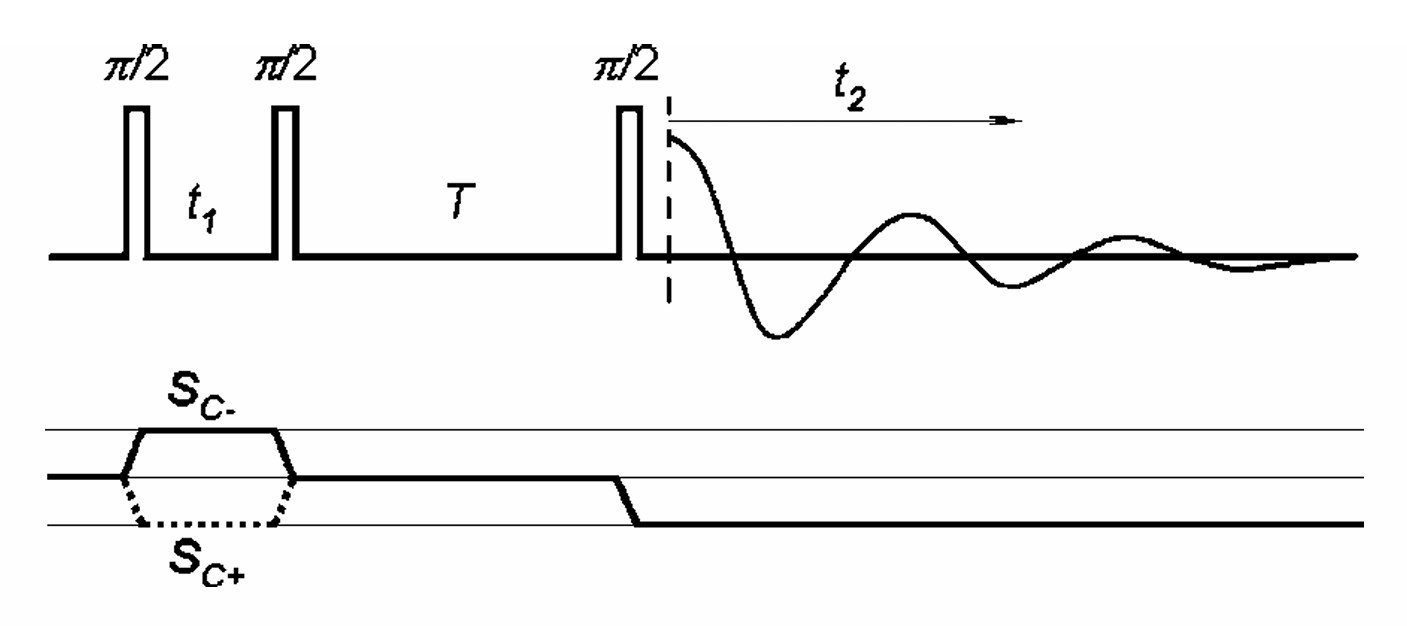
|
|
ABSTRACT: Two-dimensional (2D) Fourier transform ESR techniques, such as 2D-ELDOR, have considerably improved the resolution of ESR in studies of molecular dynamics in complex fluids such as liquid crystals and membrane vesicles and in spin labeled polymers and peptides. A well-developed theory based on the stochastic Liouville equation (SLE) has been successfully employed to analyze these experiments. However, one fundamental assumption has been utilized to simplify the complex analysis, viz. the pulses have been treated as ideal non-selective ones, which therefore provide uniform irradiation of the whole spectrum. In actual experiments, the pulses are of finite width causing deviations from the theoretical predictions, a problem that is exacerbated by experiments performed at higher frequencies. In the present paper we provide a method to deal with the full SLE including the explicit role of the molecular dynamics, the spin Hamiltonian and the radiation field during the pulse. The computations are rendered more manageable by utilizing the Trotter formula, which is adapted to handle this SLE in what we call a "Split Super-Operator" method. Examples are given for different motional regimes, which show how 2D-ELDOR spectra are affected by the finite pulse widths. The theory shows good agreement with 2D-ELDOR experiments performed as a function of pulse width.
|
|
|
EPR Distance Measurements Support a Model for Long-Range Radical Initiation in E. coli Ribonucleotide Reductase
M. Bennati, J.H. Robblee, V. Mugnaini, J. Stubbe, J.H. Freed, and P. Borbat.
J. Am. Chem. Soc. 127, 15014-15015 (2005).
<doi: 10.1021/ja054991y>
PMID:
16248626
|
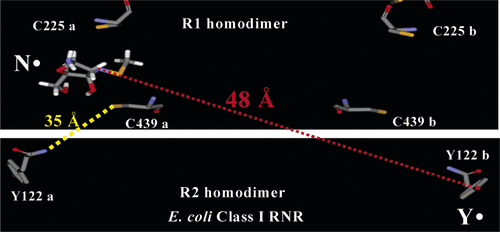
|
|
ABSTRACT: The class I E. coli ribonucleotide reductase, composed of homodimers of R1 and R2, catalyzes the conversion of nucleoside diphosphates to deoxynucleoside diphosphates. The reduction process involves the tyrosyl radical on R2 that generates a transient thiyl radical on R1 over a proposed distance of 35 Å. A mechanism-based inhibitor, 2′-azido-2′-deoxyuridine-5′-diphosphate, that reduces the tyrosyl radical on R2 and forms a nitrogen-centered radical on R1 has provided a method to measure the diagonal distance between the two subunits. PELDOR and DQC paramagnetic resonance methods give rise to a distance of 48 Å, similar to that calculated from a docking model of the R1 and R2 structures.
|
|
|
ESR and Molecular Dynamics
J.H. Freed.
In Biomedical EPR, Part B: Methodology, Instrumentation and Dynamics, S.S. Eaton, G.R. Eaton, and L.J. Berliner, Eds.
Biological Magnetic Resonance 24, Chapter 9, pp. 239-268 (2005). (Kluwer, NY).
|
|
|
ESR and Molecular Dynamics
J.H. Freed.
In Biomedical EPR, Part B: Methodology, Instrumentation and Dynamics, S.S. Eaton, G.R. Eaton, and L.J. Berliner, Eds.
Biological Magnetic Resonance 24, Chapter 9, pp. 239-268 (2005). (Kluwer, NY).
<doi: 10.1007/0-306-48533-8_9>
|
|
|
ABSTRACT: The development of ESR for the study of spin-relaxation and molecular dynamics of organic radicals and spin labels in fluids is reviewed from a historical perspective.
|
|
|
A Variable Temperature EPR Study of Mn2+-doped NH4Cl0.9I0.1 Single Crystal at 170 GHz: Zero-Field Splitting Parameter and its Absolute Sign
S.K. Misra, S.I. Andronenko, P. Chand, K.A. Earle, S.V. Paschenko, J.H. Freed.
J. Magn. Reson. 174, 265-269 (2005).
<doi: 10.1016/j.jmr.2005.02.015>
PMID:
15862243
|
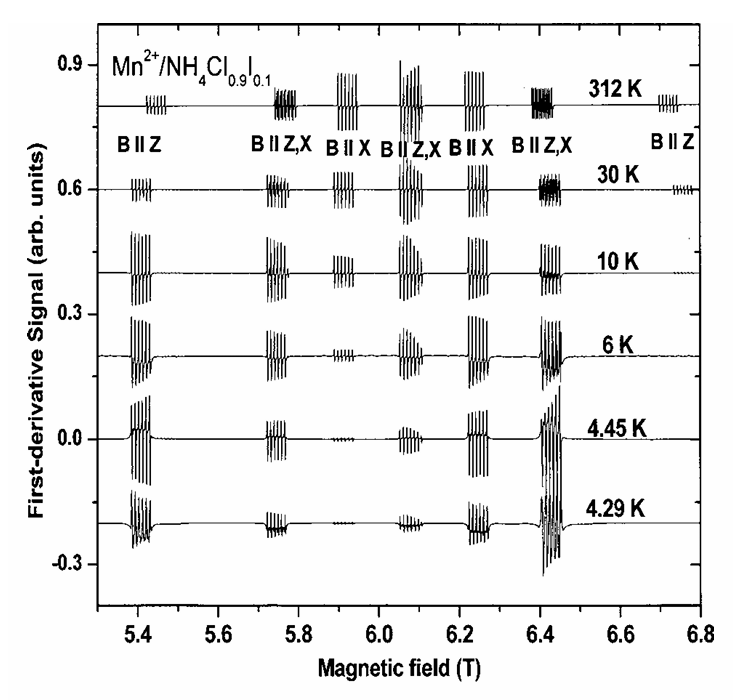
|
|
ABSTRACT: EPR measurements have been carried out on a single crystal of Mn2+-doped NH4Cl0.9I0.1 at 170-GHz in the temperature range of 312–4.2 K. The spectra have been analyzed (i) to estimate the spin-Hamiltonian parameters; (ii) to study the temperature variation of the zero-field splitting (ZFS) parameter; (iii) to confirm the negative absolute sign of the ZFS parameter unequivocally from the temperature-dependent relative intensities of hyperfine sextets at temperatures below 10 K; and (iv) to detect the occurrence of a structural phase transition at 4.35 K from the change in the structure of the EPR lines with temperature below 10 K.
|
|
|
The Determination of Pair Distance Distributions by Pulsed ESR Using Tikhonov Regularization
Y.-W. Chiang, P.P. Borbat, J.H. Freed.
J. Magn. Reson. 172, 279-295 (2005).
<doi: 10.1016/j.jmr.2004.10.012>
PMID:
15649755
|
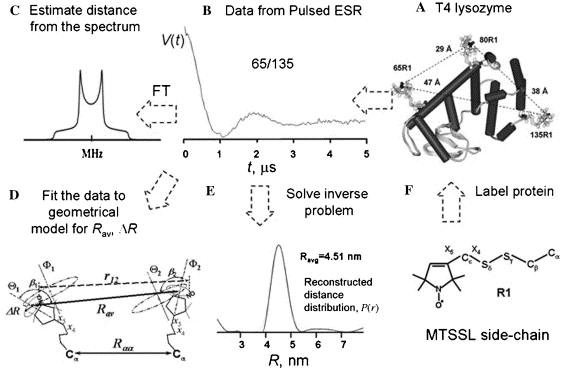
|
|
ABSTRACT: Pulsed ESR techniques with the aid of site-directed spin labeling have proven useful in providing unique structural information about proteins. The determination of distance distributions in electron spin pairs directly from the dipolar time evolution of the pulsed ESR signals by means of the Tikhonov regularization method is reported. The difficulties connected with numerically inverting this ill-posed mathematical problem are clearly illustrated. The Tikhonov regularization with the regularization parameter determined by the L-curve criterion is then described and tested to confirm its accuracy and reliability. The method is applied to recent experimental results on doubly labeled proteins that have been studied using two pulsed ESR techniques, double quantum coherence (DQC) ESR and double electron–electron resonance (DEER). The extracted distance distributions are able to provide valuable information about the conformational constraints in various partially folded states of proteins. This study supplies a mathematically reliable method for extracting pair distributions from pulsed ESR experimental data and has extended the use of pulsed ESR to provide results of greater value for structural biology.
|
|
|
New Method for Determining Tie-lines in Coexisting Membrane Phases using Spin-label ESR
Y.-W. Chiang, J. Zhao, J. Wu, Y. Shimoyama, J.H. Freed, and G.W. Feigenson.
Biochim. Biophys. Acta 1668, 99-105 (2005).
<doi: 10.1016/j.bbamem.2004.11.010>
PMID:
15670735
|
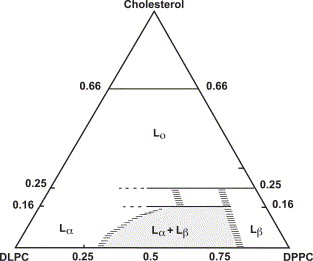
|
|
ABSTRACT: A full description of coexisting phases includes their respective compositions, which are provided by the thermodynamic tie-lines. Fluorescence microscopy enables visualization of coexisting lipid phases in giant unilamellar phases, but the composition information is missing. For cholesterol-containing lipid mixtures, knowledge of the compositions of the coexisting phases is important for understanding the nature of "membrane rafts". We propose and demonstrate a new method, based on ESR spectroscopy, for determining tie-lines in regions of two-phase coexistence in a ternary lipid mixture. Over 100 different lipid compositions containing the spin-labeled phospholipid 16-PC in or near the two-phase coexistence region of the liquid-disordered and the gel phases of dipalmitoyl-PC/dilauroyl-PC/cholesterol (DPPC/DLPC/Chol) were studied to determine five tie-lines, spread over virtually the full range of this coexistence region. The method is based on the facts that (1) along a tie-line the ESR spectrum must be a superposition of the two ESR spectra from the respective single phases at the phase boundaries (connected by the tie-line) in a ratio given by the lever rule; (2) along a tie-line the partition coefficient, Kp, for the spin-label, which is also determined in our method, must be constant. We do find that Kp for 16-PC is close to unity, but its value depends on the particular tie-line. The coexisting phases in equilibrium are characterized by the Kp of the spin-label and its respective dynamic parameters obtained from fitting the ESR spectra to dynamical models.
|
|
|
Pulsed Three-Dimensional Electron Spin Resonance Microscopy
A. Blank, C.R. Dunnam, P.P. Borbat, and J.H. Freed.
Appl. Phys. Lett. 85, 5430-5432 (2004).
<doi: 10.1063/1.1828599>
|
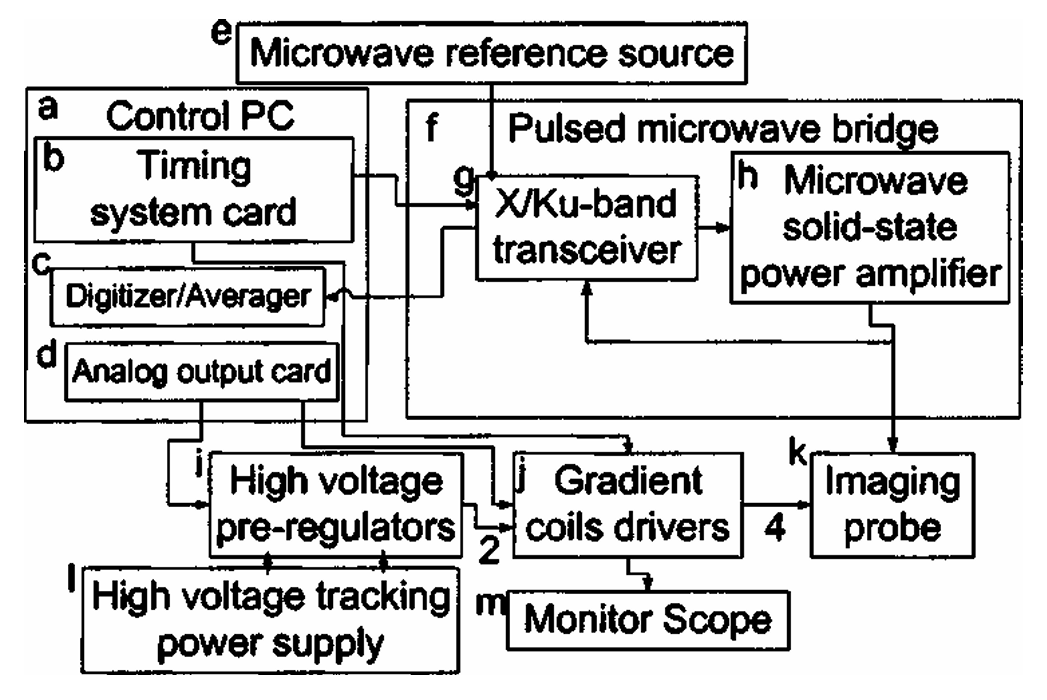
|
|
ABSTRACT: A three-dimensional (3D) electron spin resonance (ESR) microimaging system, operating in pulse mode at 9 GHz is presented. This microscope enables the acquisition of spatially resolved magnetic resonance signals of free-radicals in solid or liquid samples with a resolution of up to ˜3.5 × 7 × 11.4 μm in 20 min of acquisition. The detection sensitivity at room temperature is ˜1.2 × 109 spins / √Hz, which enables the measurement of ˜2 × 107 spins in each voxel after 60 min of acquisition. The resolution and detection sensitivity are the best obtained so far for ESR at ambient conditions of temperature and pressure. This ESR microscope can be employed in the investigation of a variety of samples in the fields of botany, life sciences, and materials science.
|
|
|
Spin-Labeled Gramicidin A: Channel Formation and Dissociation
B.G. Dzikovski, P.P. Borbat, and J.H. Freed.
Biophys. J. 87, 3504-3517 (2004).
<doi: 10.1529/biophysj.104.044305>
PMID:
15326023
PMCID:
PMC1304816
|
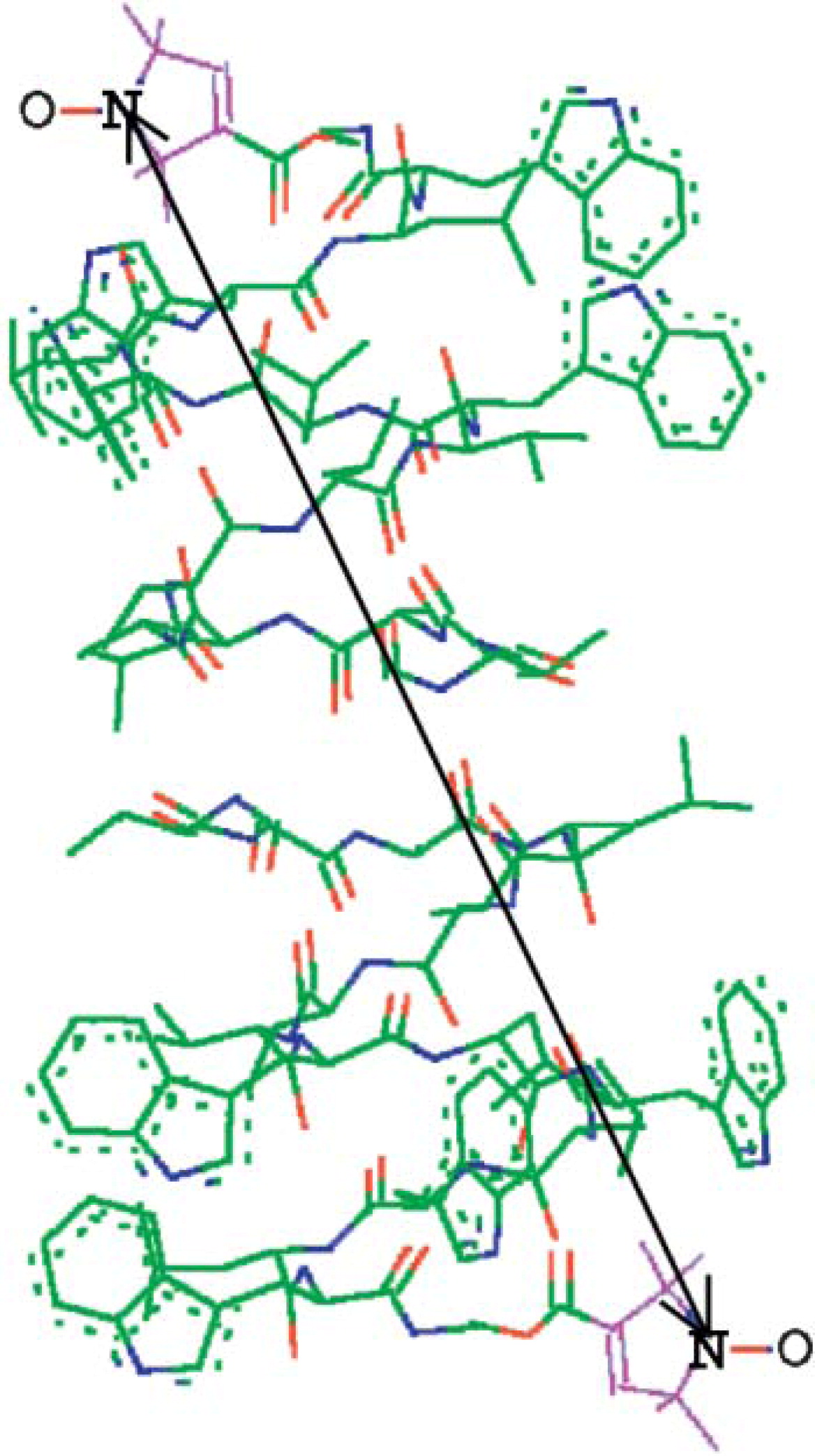
|
|
ABSTRACT: Gramicidin A was studied by continuous wave electron spin resonance (CW-ESR) and by double-quantum coherence electron spin resonance (DQC-ESR) in several lipid membranes (using samples that were macroscopically aligned by isopotential spin-dry ultracentrifugation) and vesicles. As a reporter group, the nitroxide spin-label was attached at the C-terminus yielding the spin-labeled product (GAsl). ESR spectra of aligned membranes containing GAsl show strong orientation dependence. In DPPC and DSPC membranes at room temperature the spectral shape is consistent with high ordering, which, in conjunction with the observed high polarity of the environment of the nitroxide, is interpreted in terms of the nitroxide moiety being close to the membrane surface. In contrast, spectra of GAsl in DMPC membranes indicate deeper embedding and tilt of the NO group. The GAsl spectrum in the DPPC membrane at 35°C (the gel to Pβ phase transition) exhibits sharp changes, and above this temperature becomes similar to that of DMPC. The dipolar spectrum from DQC-ESR clearly indicates the presence of pairs in DMPC membranes. This is not the case for DPPC, rapidly frozen from the gel phase; however, there are hints of aggregation. The interspin distance in the pairs is 30.9 Å, in good agreement with estimates for the head-to-head GAsl dimer (the channel-forming conformation), which matches the hydrophobic thickness of the DMPC bilayer. Both DPPC and DSPC, apparently as a result of hydrophobic mismatch between the dimer length and bilayer thickness, do not favor the channel formation in the gel phase. In the Pβ and Lα phases of DPPC (above 35°C) the channel dimer forms, as evidenced by the DQC-ESR dipolar spectrum after rapid freezing. It is associated with a lateral expansion of lipid molecules and a concomitant decrease in bilayer thickness, which reduces the hydrophobic mismatch. A comparison with studies of dimer formation by other physical techniques indicates the desirability of using low concentrations of GA (˜0.4–1 mol %) accessible to the ESR methods employed in the study, since this yields non-interacting dimer channels.
|
|
|
A Multifrequency Electron Spin Resonance Study of T4 Lysozyme Dynamics Using the Slowly Relaxing Local Structure Model
Z. Liang, Y. Lou, J.H. Freed, L. Columbus and W.L. Hubbell.
J. Phys. Chem. B 108, 17649-17659 (2004).
<doi: 10.1021/jp0484837>
|
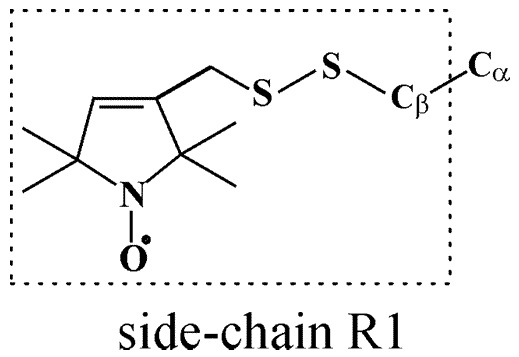
|
|
ABSTRACT: Electron spin resonance (ESR) spectra were obtained at 250 and 9 GHz for nitroxide-labeled mutants of the protein T4 lysozyme in aqueous solution over a range of temperatures from 2 to 37.5 °C. Two mutants labeled at sites 72 and 131 were studied and compared. The mutant sites are solvent exposed and free of tertiary interactions with other side chains, but the former is at the center of a 5 turn helix, whereas the latter site is on a small two and a half turn helix. The 250 GHz ESR spectra, because of their "fast time scale", are rather insensitive to the slow overall tumbling motion of the protein. Thus, they are qualitatively different for the two mutants, implying that there are different local dynamics at the two sites. The 9 GHz spectra, which are significantly affected by the overall tumbling and are less sensitive to the internal dynamics, do not show such marked differences between the two sites. The 250 and 9 GHz spectra for each mutant and temperature were simultaneously fit to the slowly relaxing local structure (SRLS) model for slow-motional ESR, using newly developed software. The SRLS model explicitly accounts for the overall tumbling of the protein and the internal modes of motion, which include the motion of the nitroxide side chain (expected to be the same for both mutants) and backbone fluctuations. Very good simultaneous fits are obtained. Whereas two conformers (or spectral components) are typically detected at the lower temperatures, only a single component is observed at the higher temperatures. The significant differences in the high-frequency spectra for the two mutants are readily attributed mainly to a difference in their respective local ordering. That is, site 72 exhibits significantly greater local ordering than does site 131, which is expected from the greater rigidity of the larger helix on which the 72 site is located. The results of this multifrequency study are compared with a previous 9 GHz study. A description of the application of the SRLS model in such a multifrequency study is provided.
|
|
|
Dynamic Molecular Structure of DPPC-DLPC-Cholesterol Ternary Lipid System by Spin-Label Electron Spin Resonance
Y.-W. Chiang, Y. Shimoyama, G.W. Feigenson, and J.H. Freed.
Biophys. J. 87, 2483-2496 (2004).
<doi: 10.1529/biophysj.104.044438>
PMID:
15454445
PMCID:
PMC1304668
|
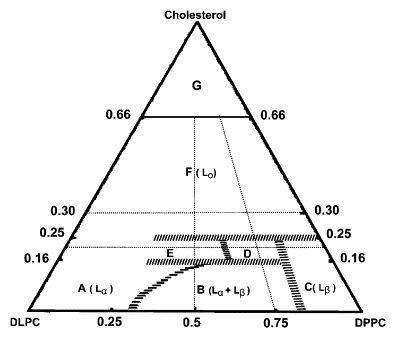
|
|
ABSTRACT: The hydrated ternary lamellar lipid mixture of dipalmitoyl-PC/dilauroyl-PC/cholesterol (DPPC/DLPC/Chol) has been studied by electron spin resonance (ESR) to reveal the dynamic structure on a molecular level of the different phases that exist and coexist over virtually the full range of composition. The spectra for more than 100 different compositions at room temperature were analyzed by nonlinear least-squares fitting to provide the rotational diffusion rates and order parameters of the end-chain labeled phospholipid 16-PC. The ESR spectra exhibit substantial variation as a function of composition, even though the respective phases generally differ rather modestly from each other. The Lα and Lβ phases are clearly distinguished, with the former exhibiting substantially lower ordering and greater motional rates, whereas the well-defined LOphase exhibits the greatest ordering and relatively fast motional rates. Typically, smaller variations occur within a given phase. The ESR spectral analysis also yields phase boundaries and coexistence regions which are found to be consistent with previous results from fluorescence methods, although new features are found. Phase coexistence regions were in some cases confirmed by observing the existence of isosbestic points in the absorption mode ESR spectra from the phases. The dynamic structural properties of the DPPC-rich Lβ and DLPC-rich Lα phases, within their two-phase coexistence region do not change with composition along a tie-line, but the ratio of the two phases follows the lever rule in accordance with thermodynamic principles. The analysis shows that 16-PC spin-label partitions nearly equally between the Lα and Lβ phases, making it a useful probe for studying such coexisting phases. Extensive study of two-phase coexistence regions requires the determination of tie-lines, which were approximated in this study. However, a method is suggested to accurately determine the tie-lines by ESR.
|
|
|
A Three-Dimensional Electron Spin Resonance Microscope
A. Blank, C.R. Dunnam, P.P. Borbat, and J.H. Freed.
Rev. Sci. Instrum. 75, 3050-3061 (2004).
<doi: 10.1063/1.1786353>
|
|
|
ABSTRACT: An electron spin resonance (ESR) imaging system, capable of acquiring three-dimensional (3D) images with a resolution of ˜10 × 10 × 30 μm in a few minutes of acquisition, is presented. This ESR microscope employs a commercial continuous wave ESR spectrometer, working at 9.1 GHz, in conjunction with a miniature imaging probe (resonator + gradient coils), gradient current drivers, and control software. The system can acquire the image of a small (˜ 1.5 × 1.5 × 0.25 mm) sample either by the modulated field gradient method, the projection reconstruction method, or by a combination of the two. A short discussion regarding the resolution of the modulated field gradient method in two-dimensional (2D) and 3D imaging is given. Detailed descriptions of the various system components are provided, along with several examples of 2D and 3D images that demonstrate the capabilities of the system.
|
|
|
Enrichment of Endoplasmic Reticulum with Cholesterol Inhibits Sarcoplasmic-Endoplasmic Reticulum Calcium ATPase-2b Activity in Parallel with Increased Order of Membrane Lipids
Y. Li, M. Ge, L. Ciani, G. Kuriakose, E.J. Westover, M. Dura, D.F. Covey, J.H. Freed, F.R. Maxfield, J. Lytton, and I. Tabas.
J. Biol. Chem. 279, 37030-37039 (2004).
|
|
|
Enrichment of Endoplasmic Reticulum with Cholesterol Inhibits Sarcoplasmic-Endoplasmic Reticulum Calcium ATPase-2b Activity in Parallel with Increased Order of Membrane Lipids
Y. Li, M. Ge, L. Ciani, G. Kuriakose, E.J. Westover, M. Dura, D.F. Covey, J.H. Freed, F.R. Maxfield, J. Lytton, and I. Tabas.
J. Biol. Chem. 279, 37030-37039 (2004).
<doi: 10.1074/jbc.M405195200>
PMID:
15215242
|
|
|
ABSTRACT: Macrophages in advanced atherosclerotic lesions accumulate large amounts of unesterified, or "free," cholesterol (FC). FC accumulation induces macrophage apoptosis, which likely contributes to plaque destabilization. Apoptosis is triggered by the enrichment of the endoplasmic reticulum (ER) with FC, resulting in depletion of ER calcium stores, and induction of the unfolded protein response. To explain the mechanism of ER calcium depletion, we hypothesized that FC enrichment of the normally cholesterol-poor ER membrane inhibits the macrophage ER calcium pump, sarcoplasmic-endoplasmic reticulum calcium ATPase-2b (SERCA2b). FC enrichment of ER membranes to a level similar to that occuring in vivo inhibited both the ATPase activity and calcium sequestration function of SERCA2b. Enrichment of ER with ent-cholesterol or 14:0-18:0 phosphatidylcholine, which possess the membrane-ordering properties of cholesterol, also inhibited SERCA2b. Moreover, at various levels of FC enrichment of ER membranes, there was a very close correlation between increasing membrane lipid order, as monitored by 16-doxyl-phosphatidycholine electron spin resonance, and SERCA2b inhibition. In view of these data, we speculate that SERCA2b, a conformationally active protein with 11 membrane-spanning regions, loses function due to decreased conformational freedom in FC-ordered membranes. This biophysical model may underlie the critical connection between excess cholesterol, unfolded protein response induction, macrophage death, and plaque destabilization in advanced atherosclerosis.
|
|
|
High Field ESR: Applications to Protein Structure and Dynamics: HF ESR Protein Structure and Dynamics
K.A. Earle and A.I. Smirnov.
In Very High Frequency (VHF) ESR/EPR, O. Grinberg and L.J. Berliner, Eds.
Biological Magnetic Resonance 22, Chapter 4, pp. 95-143 (2004). (Kluwer, NY).
<doi: 10.1007/978-1-4757-4379-1_4>
|
|
|
ABSTRACT: Electron Spin Resonance at high magnetic fields (HF ESR) is rapidly developing into a powerful biophysical tool which is uniquely positioned to address complex aspects of structure and dynamics of proteins, membranes, and macromolecular assemblies at molecular level. The source of the contemporary resurgence of interest in ESR as a biophysical tool is that there are, broadly speaking, three large groups of problems, which can be approached with ESR methods but cannot easily be studied by traditional structural techniques: (1) structure and dynamics of large molecular weight proteins in solution; (2) membrane and membrane-associated proteins: structure, location with respect to the membrane, side-chain dynamics, and interactions with other membrane components or DNA's and RNA's; (3) fast conformational transitions of proteins and RNA's in solution, protein folding and re-folding. The focus of this review is mainly on HF ESR spin-labeling techniques, primarily via nitroxide spin labels, as this method is the most flexible and is even applicable to proteins which are otherwise ESR-silent. We start with physical aspects of ESR of nitroxide spin labels at high magnetic fields in order to categorize the characteristic information that can be gained from such experiments. Then we describe practical applications of spin-labeling HF ESR to study structure and dynamics of complex biophysical systems. We also discuss several details of the ESR motional theory based on the stochastic Liouville equation (SLE) that are relevant to nitroxide line shape analysis in order to appreciate both the information available from and the limitations of the method. Particular emphasis is given to multifrequency HF ESR methods in studies of spin-labeled membranes and biopolymers. Finally, we review the use of HF ESR in applications to molecular structure including distance measurements, determination of molecular orientations from ESR of ordered samples, and structural studies based on g-factor measurements.
|
|
|
The Development of High Field/High Frequency ESR: Historical Overview
J.H. Freed.
In Very High Frequency (VHF) ESR/EPR, O. Grinberg and L.J. Berliner, Eds.
Biological Magnetic Resonance 22, Chapter 2, pp. 19-43 (2004). (Kluwer, NY).
<doi: 10.1007/978-1-4757-4379-1_2>
|
|
|
ABSTRACT: We discuss the development of high field ESR into a powerful and flexible tool for studies of structure and dynamics in a wide variety of systems including those of biological interest. A range of techniques are discussed with particular emphasis on the developments at Cornell University, but the contributions of other groups to the constant refinement of the state of the art are also noted.
|
|
|
|
|
Measurement of Large Distances in Biomolecules Using Double-Quantum Filtered Refocused Electron Spin-Echoes
P.P. Borbat, J.H. Davis, S.E. Butcher, and J.H. Freed.
J. Am. Chem. Soc. 126, 7746-7747 (2004).
<doi: 10.1021/ja049372o>
PMID:
15212500
|
|
|
ABSTRACT: It is shown how the new technique of double-quantum filtered refocused electron spin–echoes is a significant improvement over double-quantum coherence ESR, since it increases the experimental acquisition time. This enables the measurement of longer distances in bilabeled biomolecules. The method is demonstrated on a long double-stranded A-type RNA, spin labeled at both ends. The measured distance of 72 Å is in excellent agreement with molecular modeling.
|
|
|
High-Power 95 GHz Pulsed Electron Spin Resonance Spectrometer
W. Hofbauer, K.A. Earle, C.R. Dunnam, J.K. Moscicki, and J.H. Freed.
Rev. Sci. Instrum. 75, 1194-1208 (2004).
<doi: 10.1063/1.1710700>
|
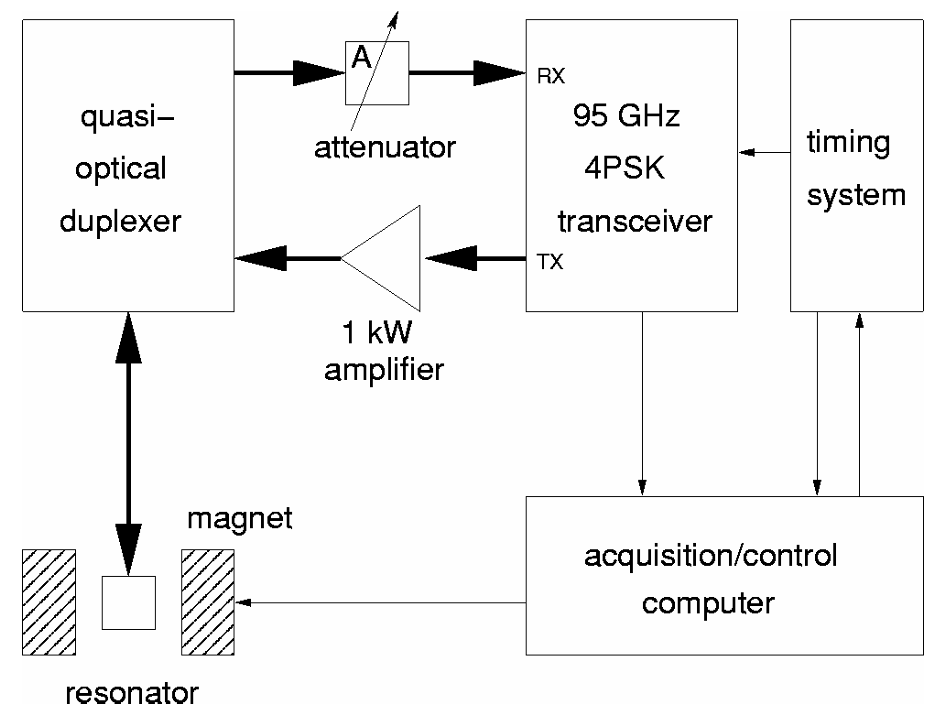
|
|
ABSTRACT: High-field/high-frequency electron spin resonance (ESR) offers improved sensitivity and resolution compared to ESR at conventional fields and frequencies. However, most high-field/high-frequency ESR spectrometers suffer from limited mm-wave power, thereby requiring long mm-wave pulses. This precludes their use when relaxation times are short, e.g., in fluid samples. Low mm-wave power is also a major factor limiting the achievable spectral coverage and thereby the multiplex advantage of Fourier transform ESR (FTESR) experiments. High-power pulses are needed to perform two-dimensional (2D) FTESR experiments, which can unravel the dynamics of a spin system in great detail, making it an excellent tool for studying spin and molecular dynamics. We report on the design and implementation of a high-power, high-bandwidth, pulsed ESR spectrometer operating at 95 GHz. One of the principal design goals was the ability to investigate dynamic processes in aqueous samples at physiological temperatures with the intent to study biological systems. In initial experiments on aqueous samples at room temperature, we achieved 200 MHz spectral coverage at a sensitivity of 1.1×1010√(s) spins and a dead time of less than 50 ns. 2D-electron-electron double resonance experiments on aqueous samples are discussed to demonstrate the practical application of such a spectrometer.
|
|
|
Hydration, Structure, and Molecular Interactions in the Headgroup Region of Dioleoylphosphatidyl-Choline Bilayers: An Electron Spin Resonance Study
M. Ge and J.H. Freed.
Biophys. J. 85, 4023-40. (2003).
<doi: 10.1016/S0006-3495(03)74816-4>
PMID:
14645091
PMCID:
PMC1303703
|
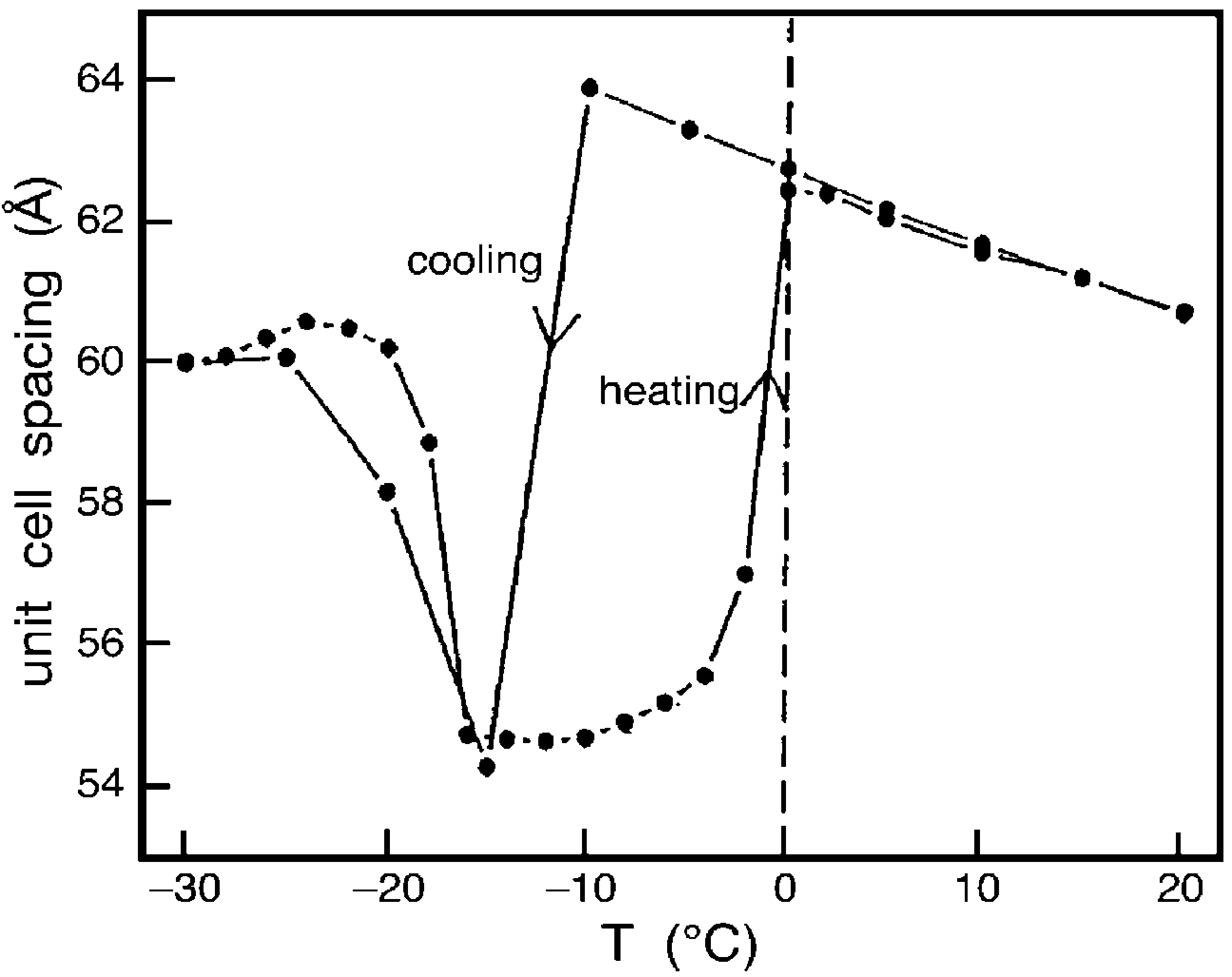
|
|
ABSTRACT: The relationship between bilayer hydration and the dynamic structure of headgroups and interbilayer water in multilamellar vesicles is investigated by electron spin resonance methods. Temperature variations of the order parameter of a headgroup spin label DPP-Tempo in DOPC in excess water and partially dehydrated (10wt % water) show a cusp-like pattern around the main phase transition, Tc. This pattern is similar to those of temperature variations of the quadrupolar splitting of interbilayer D2O in PC and PE bilayers previously measured by 2H NMR, indicating that the ordering of the headgroup and the interbilayer water are correlated. The cusp-like pattern of these and other physical properties around Tc are suggestive of quasicritical fluctuations. Also, an increase (a decrease) in ordering of DPP-Tempo is correlated with water moving out of (into) interbilayer region into (from) the bulk water phase near the freezing point, Tf. Addition of cholesterol lowers Tf, which remains the point of increasing headgroup ordering. Using the small water-soluble spin probe 4-PT, it is shown that the ordering of interbilayer water increases with bilayer dehydration. It is suggested that increased ordering in the interbilayer region, implying a lowering of entropy, will itself lead to further dehydration of the interbilayer region until its lowered pressure resists further flow, i.e., an osmotic phenomenon.
|
|
|
High Resolution Electron Spin Resonance Microscopy
A. Blank, C.R. Dunnam, P.P. Borbat, and J.H. Freed.
J. Magn. Reson. 165, 116-127 (2003).
<doi: 10.1016/S1090-7807(03)00254-4>
PMID:
14568522
|
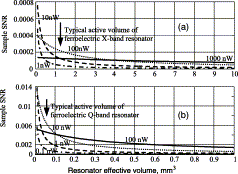
|
|
ABSTRACT: NMR microscopy is routinely employed in fields of science such as biology, botany, and materials science to observe magnetic parameters and transport phenomena in small scale structures. Despite extensive efforts, the resolution of this method is limited (>10 μm for short acquisition times), and thus cannot answer many key questions in these fields. We show, through theoretical prediction and initial experiments, that ESR microscopy, although much less developed, can improve upon the resolution limits of NMR, and successfully undertake the 1 μm resolution challenge. Our theoretical predictions demonstrate that existing ESR technology, along with advanced imaging probe design (resonator and gradient coils), using solutions of narrow linewidth radicals (the trityl family), should yield 64 × 64 pixels 2D images (with z slice selection) with a resolution of 1 × 1 × 10 μm at ˜60 GHz in less than 1 h of acquisition. Our initial imaging results, conducted by CW ESR at X-band, support these theoretical predictions and already improve upon the previously reported state-of-the-art for 2D ESR image resolution achieving ˜10 × 10 μm, in just several minutes of acquisition time. We analyze how future progress, which includes improved resonators, increased frequency of measurement, and advanced pulsed techniques, should achieve the goal of micron resolution.
|
|
|
Ordered and Disordered Phases Coexist in Plasma Membrane Vesicles of RBL-2H3 Mast Cells. An ESR Study
M. Ge, A. Gidwani, H.A. Brown, D. Holowka, B. Baird, and J.H. Freed.
Biophys. J. 85, 1278-1288 (2003).
<doi: 10.1016/S0006-3495(03)74563-9>
PMID:
12885671
PMCID:
PMC1303245
|
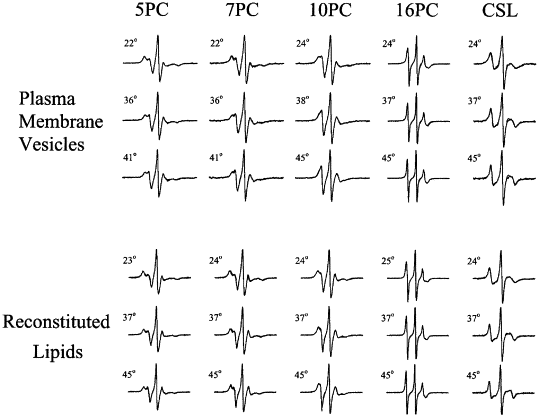
|
|
ABSTRACT: Four chain spin labels and a spin-labeled cholestane were used to study the dynamic structure of plasma membrane vesicles (PMV) prepared from RBL-2H3 mast cells at temperatures ranging from 22°C to 45°C. Analysis shows that the spectra from most labels consist of two components. The abundant spectral components exhibit substantial ordering that is intermediate between that of a liquid-ordered (Lo) phase, and that of a liquid-crystalline (Lc) phase as represented by model membranes. Also, rotational diffusion rates of the spin labels are comparable to those in the Lo phase. In contrast, the ordering for the less abundant components is much lower. These results indicate that a Lo-like region or phase (the abundant component) and an Lc-like region or phase (the less abundant component) coexist in the PMV. In contrast, membranes reconstituted from extracted lipids exhibit the more ordered phase only. This suggests that membrane-associated proteins are important for the coexistence of Lo-like and Lc-like regions in the plasma membrane. In addition, binding of the myristoylated protein, ARF6 to PMV, leads to a new spectral component for a headgroup lipid spin label that indicates the formation of plasma membrane defects by this low molecular weight GTPase.
|
|
|
Mode-Coupling SRLS versus Mode-Decoupled Model-Free N-H Bond Dynamics: Mode-Mixing and Renormalization
E. Meirovitch, Y.E. Shapiro, Z. Liang, and J.H. Freed.
J. Phys. Chem. B 107, 9898-9904 (2003).
<doi: 10.1021/jp030502>
|
|
|
ABSTRACT: The common approach to N−H motion in proteins is model-free (MF), where the global (RC) and local (RL) motions are assumed decoupled. We have recently applied to N−H bond dynamics the slowly relaxing local structure (SRLS) model, which accounts rigorously for mode-coupling. The original and extended MF formulas are perturbational expansions of SRLS with respect to the local ordering, (S02)2, when RL ≫ RC. Their functional form, number of terms equal to the number of dynamic modes, is implied by mode-decoupling, and the free diffusion eigenvalue, 1∕τ = 6RL, by the absence of strong-potential-induced renormalization. However, for N−H motion, (S02)2 is high and in the extended MF regime RL⊥ ≈ RC. Although the functional form of the original MF formula is largely valid for RC∕RL ≤ 0.01 and (S02)2 ≥ 0.8, τe MF represents the significantly reduced potential-dependent renormalized value of τ. Hence, the application of this formula to calculate NMR variables is appropriate in this parameter range, but associating τe with the local motion correlation time is inappropriate. Means to derive τ from τe are provided. For a cosine squared potential, the cone-model-based MF formula that relates τe to τ can also be used. Deriving τ from τe is important for proper characterization of the site-specific local motion and in the context of τ-dependent MF functionalities. Mode-coupling dominates the extended MF regime where SRLS must be invariably used. Eigenmode and spectral density analysis is provided in this study for the two parameter ranges associated with N−H bond motion.
|
|
|
Mode-Coupling Analysis of 15N CSA-15N-1H Dipolar Cross-Correlation in Proteins. Rhombic Potentials at the N-H Bond
E. Meirovitch, Y.E. Shapiro, V. Tugarinov, Z. Liang, and J.H. Freed.
J. Phys. Chem. B 107, 9883-9897 (2003).
<doi: 10.1021/jp030501h>
|
|
|
ABSTRACT: 15N CSA−15N-1H dipolar cross-correlation (ηxy) in proteins has been treated thus far with the model-free (MF) approach, where the global (RC) and local (RL) motions are assumed to be decoupled as a consequence of RL ≫ RC. In the context of ηxy, it is additionally assumed that the local motion is very fast and highly symmetrical. We have recently applied to auto-correlated 15N spin relaxation the slowly relaxing local structure (SRLS) approach, which accounts rigorously for mode-coupling. SRLS can analyze ηxy for arbitrary time scale separations between RL and RC. Simulations of ηxy for slow local motions are presented herein for the first time. Experimental ηxy values of RNase and AKeco could not be reproduced from best-fit parameters generated by data fitting that used axial potentials. Calculations showed they are reproducible using rhombic coupling/ordering potentials. The conformational exchange term, Rex, "absorbs" potential rhombicity when axial potentials are used to fit the data. 15N CSA variability and RC anisotropy are shown to have a small effect on the analysis. The shape of the rhombic potentials detected corresponds to nearly "planar YMXM ordering" (M denotes the local ordering frame). This potential form is consistent with the known geometry of the peptide plane, the SRLS dynamic model in the RL⊥ ≈ RC regime, and makes possible associating the local director with the Ci−1α-Ciα axis. It is shown that back-calculation of NMR variables from the best-fit parameters and comparison with the experimental counterparts is an effective tool for identifying inappropriate elements of the dynamic model and can thereby help improve it.
|
|
|
Lipid-Gramicidin Interactions: Dynamic Structure of the Boundary Lipid by 2D-ELDOR
A.J. Costa-Filho, R.H. Crepeau, P.P. Borbat, M. Ge, and J. H. Freed.
Biophys. J. 84, 3364-3378 (2003).
<doi: 10.1016/S0006-3495(03)70060-5>
PMID:
12719265
PMCID:
PMC1302896
|
|
|
ABSTRACT: The use of 2D-electron-electron double resonance (2D-ELDOR) for the characterization of the boundary lipid in membrane vesicles of DPPC and gramicidin A′ (GA) is reported. We show that 2D-ELDOR, with its enhanced spectral resolution to dynamic structure as compared with continuous-wave electron spin resonance, provides a reliable and useful way of studying lipid-protein interactions. The 2D-ELDOR spectra of the end-chain spin label 16-PC in DPPC∕GA vesicles is composed of two components, which are assigned to the bulk lipids (with sharp auto peaks and crosspeaks) and to the boundary lipids (with broad auto peaks). Their distinction is clearest for higher temperatures and higher GA concentrations. The quantitative analysis of these spectra shows relatively faster motions and very low ordering for the end chain of the bulk lipids, whereas the boundary lipids show very high "y-ordering" and slower motions. The y-ordering represents a dynamic bending at the end of the boundary lipid acyl chain, which can then coat the GA molecules. These results are consistent with the previous studies by Ge and Freed (1999) using continuous-wave electron spin resonance, thereby supporting their model for GA aggregation and HII phase formation for high GA concentrations. Improved instrumental and simulation methods have been employed.
|
|
|
A 2D-ELDOR Study of the Liquid Ordered Phase in Multilamellar Vesicle Membranes
A.J. Costa-Filho, Y. Shimoyama, and J.H. Freed.
Biophys. J. 84, 2619-2633, (2003).
<doi: 10.1016/S0006-3495(03)75067-X>
PMID:
12668470
PMCID:
PMC1302828
|
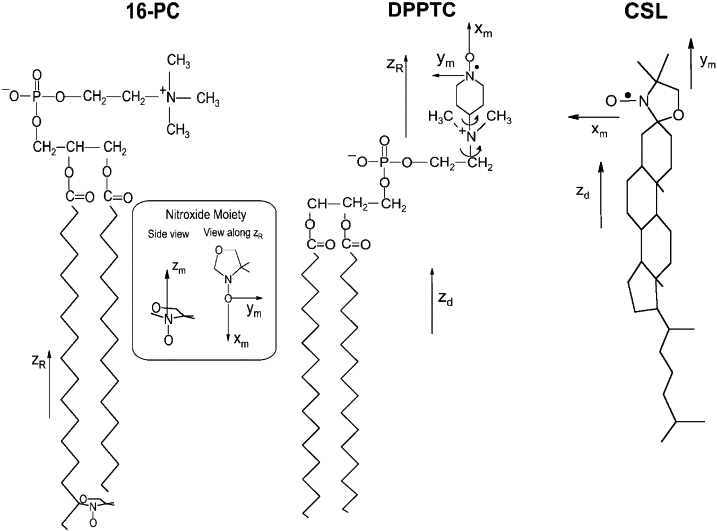
|
|
ABSTRACT: 2D-ELDOR spectroscopy has been employed to study the dynamic structure of the liquid-ordered (Lo) phase versus that of the liquid-crystalline (Lc) phase in multibilayer phospholipid vesicles without (Lc) and with (Lo) cholesterol, using end-chain and headgroup labels and spin-labeled cholestane. The spectra are in most cases found to be dramatically different for these two phases. Thus, visual inspection of the 2D-ELDOR spectra provides a convenient way to distinguish the two phases in membranes. Detailed analysis shows these observations are due to increased ordering in the Lo phase and modified reorientation rates. In the Lo phase, acyl chains undergo a faster rotational diffusion and higher ordering than in the Lc phase, whereas spin-labeled cholestane exhibits slower rotational diffusion and higher ordering. On the other hand, the choline headgroup in the Lo phase exhibits faster motion and reduced but realigned ordering versus the Lc phase. The microscopic translational diffusion rates in the Lo phase are significantly reduced in the presence of cholesterol. These results are compared with previous studies, and a consistent model is provided for interpreting them in terms of the differences in the dynamic structure of the Lo and Lc phases.
|
|
|
End-to-End Correlation for a C-12 Hydrocarbon Chain
S. Wagner, A.A. Nevzorov, J.H. Freed, and R.G. Bryant.
J. Magn. Reson. 160, 161-165 (2003)
<doi: 10.1016/S1090-7807(02)00143-X>
PMID:
12615159
|
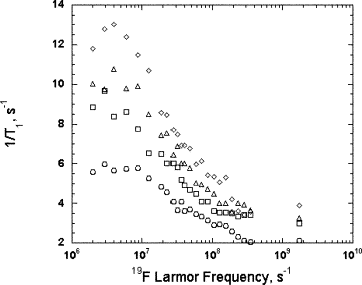
|
|
ABSTRACT: The 19F nuclear spin-lattice relaxation rate constants were measured as a function of magnetic field strength for 1,12-diaminododecane labeled at one end with a nitroxide radical and at the other with a trifluoromethyl group. The magnetic relaxation dispersion profile (MRD) reports the spectral density function appropriate to the end-to-end correlation function for the doubly labeled molecule. After extrapolation to zero concentration to eliminate the intermolecular relaxation contribution to relaxation, the resulting intramolecular MRD profile was compared with several model approaches. The rotational model for the spectral density functions as included in the Solomon–Bloembergen–Morgan equations does not describe the data well. The earlier model of Freed for nuclear spin relaxation induced by a freely diffusing paramagnetic co-solute is not rigorous for this case because the paramagnet is tethered to the observed nuclear spin and only a restricted space in the immediate vicinity of the nuclear spin is accessible for pseudo-translational diffusion of one end of the molecule with respect to the other. A generalization of the Torrey model for magnetic relaxation by translational diffusion developed by Nevzorov and Freed, which includes the effect of restrictions imposed by the finite length of the chain, describes the experiment within experimental errors. A simple modification of the Hwang–Freed model that does not specifically include the dynamical effects of the finite tether also provides a good approximation to the data when the tether chain is sufficiently long.
|
|
|
Variable-Frequency EPR Study of Mn2+-Doped NH4Cl0.9I0.1 Single Crystal at 9.6, 36, and 249.9 GHz: Structural Phase Transition
S.K. Misra, S.I. Andronenko, G. Rinaldi, P. Chand, K.A. Earle, and J.H. Freed.
J. Magn. Reson. 160, 131-138, (2003).
<doi: 10.1016/S1090-7807(02)00133-7>
PMID:
12615154
|
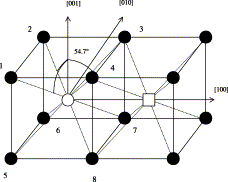
|
|
ABSTRACT: Multifrequency electron paramagnetic resonance studies on the Mn2+ impurity ion in a mixed single crystal NH4Cl0.9I0.1 were carried out at 9.62 (X-band) in the range 120–295 K, at 35.87 (Q-band) at 77 and 295 K, and at 249.9 GHz (far-infrared band) at 253 K. The high-field EPR spectra at 249.9 GHz are well into the high-field limit leading to a considerable simplification of the spectra and their interpretation. Three magnetically inequivalent, but physically equivalent, Mn2+ ions with their respective magnetic Z-axes oriented along the crystallographic [1 0 0], [0 1 0], [0 0 1] axes were observed. Simultaneous fitting of EPR line positions observed at X-, Q-, and far infra-red bands was performed using a least-squares procedure and matrix diagonalization to estimate accurately the Mn2+ spin-Hamiltonian parameters. The temperature variation of the linewidth and peak-to-peak intensities of the EPR lines indicate the presence of λ-transitions in the mixed NH4Cl0.9I0.1 crystal at 242 and 228 K consistent with those observed in the pure NH4Cl and NH4I crystals, respectively. A superposition-model analysis of the spin-Hamiltonian parameters reveals that the local environment of the Mn2+ ion is considerably reorganized to produce axially symmetric crystal fields about the respective Z-axes of the three magnetically inequivalent ions as a consequence of the vacancy created due to charge-compensation when the divalent Mn2+ ion substitutes for a monovalent NH4+ ion in the NH4Cl0.9I0.1 crystal. This reorganization is almost the same as that observed in NH4Cl and NH4I single crystals, although the latter two are characterized by different, simple cubic and face-centered cubic, structures.
|
|
|
Phase Relaxation in a Many-Body System of Diffusing Spins: Slow Motional Limit
A.A. Nevzorov and J.H. Freed.
J. Chem. Phys. 117, 282-287 (2002).
<doi: 10.1063/1.1481764>
|
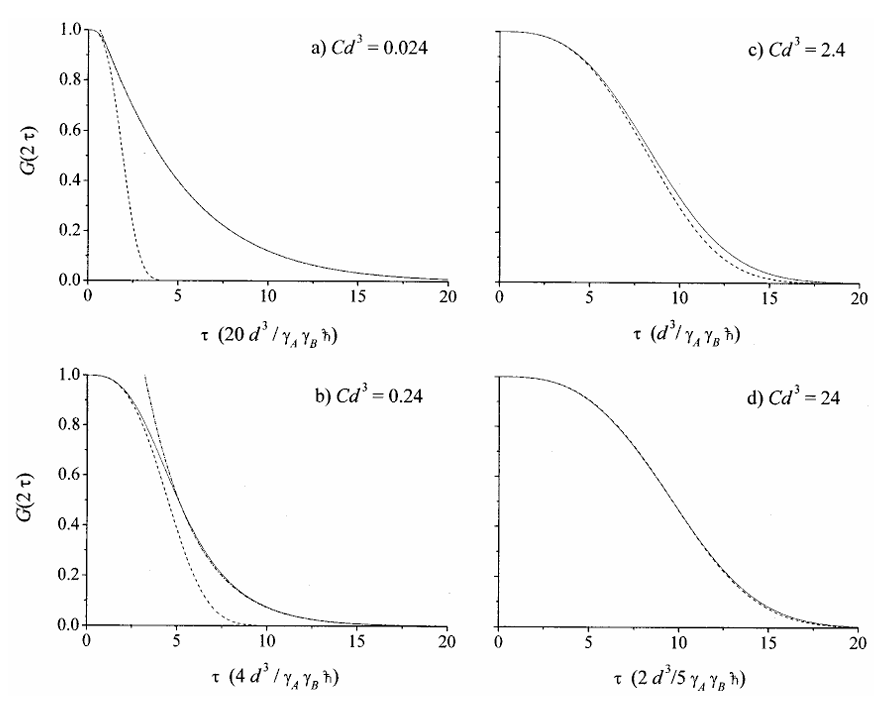
|
|
ABSTRACT: The echo amplitude decay of a diffusing electron-spin-bearing molecule interacting with a diffusing many-spin bath of proton-containing molecules has been studied theoretically in the slow motional limit. Closed-form asymptotic expressions for the short- and long-time behavior of the echo envelope have been obtained. In contrast to the well-studied fast-motional limit, the echo envelope cannot be described by a simple exponential decay, and its rate exhibits a D1∕3T dependence on the relative translational diffusion coefficient, DT For low proton concentrations and long pulse delay times, τ, an exponential τ9∕8-decay of the echo amplitude is found, whereas for high proton concentrations and short τ's the decay exhibits an exponential τ3-behavior. These limiting analytical results are compared with the exact numerical solutions to establish their range of validity.
|
|
|
Modern ESR Methods in Studies of the Dynamic Structure of Proteins and Membranes
J.H. Freed.
In EPR in the 2lst. Century , A. Kawamori, J. Yamauchi, and H. Ohta, Eds.
Elsevier Science, Amsterdam, Netherlands, 719-730 (2002).
<doi: 10.1016/B978-044450973-4/50117-7>
|
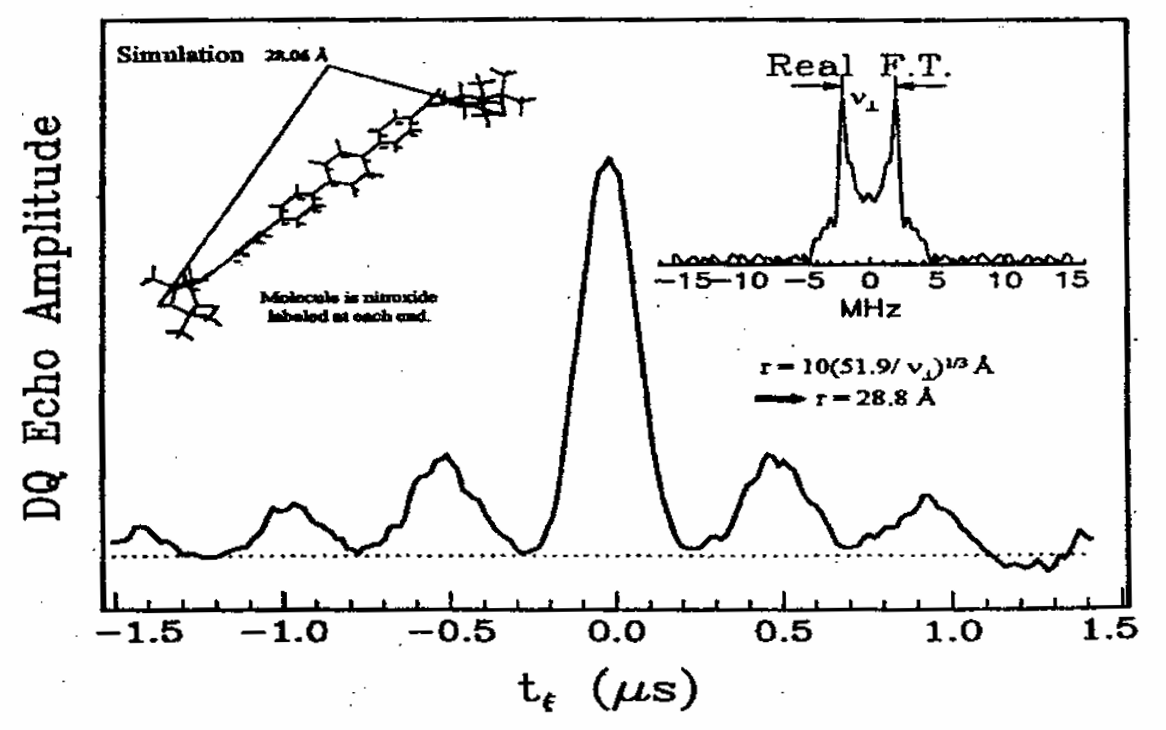
|
|
ABSTRACT: A review of modern electron spin resonance (ESR) techniques for studying the dynamic structure of proteins and membranes using nitroxide spin labels is presented. In particular multiple quantum coherence FT-ESR, multifrequency ESR based on high frequency ESR and two dimensional ESR to study the structure and dynamics of bio-membranes and proteins are illustrated by several examples. Distances in biomolecules have been determined accurately. The details of complex dynamics in proteins and the dynamic structure of membranes were characterized.
|
|
|
Domain Flexibility in Ligand-Free and Inhibitor-Bound Escherichia coli Adenylate Kinase Based on a Mode-Coupling Analysis of 15N Spin Relaxation
Y.E. Shapiro, E. Kahana, V. Tugarinov, Z. Liang, J.H. Freed, and E. Meirovitch.
Biochemistry 41, 6271-6281 (2002).
Supporting Information
<doi: 10.1021/bi012132q>
PMID:
12009888
|
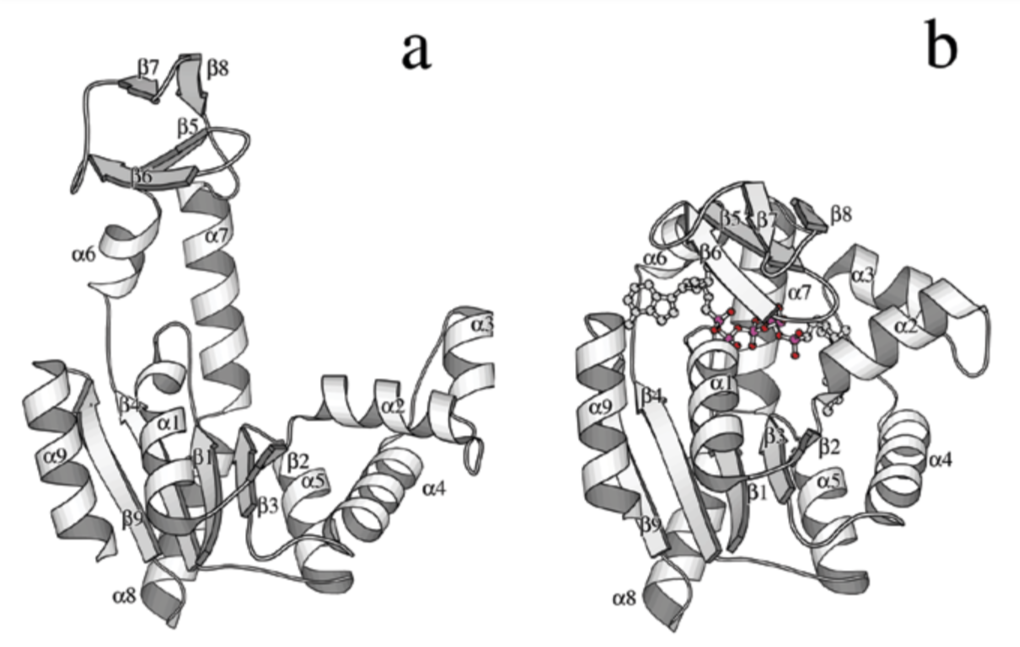
|
|
ABSTRACT: Adenylate kinase from Escherichia coli (AKeco), consisting of a 23.6-kDa polypeptide chain folded into domains CORE, AMPbd, and LID catalyzes the reaction AMP + ATP ↔ 2ADP. The domains AMPbd and LID execute large-amplitude movements during catalysis. Backbone dynamics of ligand-free and AP5A-inhibitor-bound AKeco is studied with slowly relaxing local structure (SRLS) 15N relaxation, an approach particularly suited when the global (τm) and the local (τ) motions are likely to be coupled. For AKeco τm = 15.1 ns, whereas for AKeco*AP5A τm = 11.6 ns. The CORE domain of AKeco features an average squared order parameter, <S2>, of 0.84 and correlation times τf = 5−130 ps. Most of the AKeco*AP5A backbone features <S2> = 0.90 and τf = 33−193 ps. These data are indicative of relative rigidity. Domains AMPbd and LID of AKeco, and loops β1∕α1, α2∕α3, α4∕β3, α5∕β4, and β8∕α7 of AKeco*AP5A, feature a novel type of protein flexibility consisting of nanosecond peptide plane reorientation about the Ci−1α−Ciα axis, with correlation time τ⊥ = 5.6−11.3 ns. The other microdynamic parameters underlying this dynamic model include S2 = 0.13−0.5, τ∥ on the ps time scale, and a diffusion tilt βMD ranging from 12 to 21°. For the ligand-free enzyme the τ⊥ mode was shown to represent segmental domain motion, accompanied by conformational exchange contributions Rex ≤ 4.4 s-1. Loop α4∕β3 and α5∕β4 dynamics in AKeco*AP5A is related to the "energetic counter-balancing of substrate binding" effect apparently driving kinase catalysis. The other flexible AKeco*AP5A loops may relate to domain motion toward product release.
|
|
|
Protein Structure Determination Using Long-Distance Constraints from Double-Quantum Coherence ESR: Study of T4 Lysozyme
P.P. Borbat, H.S. Mchaourab, and J.H. Freed.
J. Am. Chem. Soc. 124, 5304-5314 (2002).
<doi: 10.1021/ja020040y>
PMID:
11996571
|
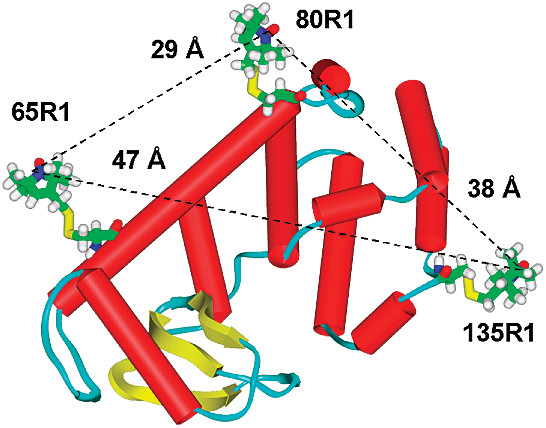
|
|
ABSTRACT: We report the use of a novel pulsed ESR technique for distance measurement, based on the detection of double quantum coherence (DQC), which yields high quality dipolar spectra, to significantly extend the range of measurable distances in proteins using nitroxide spin-labels. Eight T4 lysozyme (T4L) mutants, doubly labeled with methanethiosulfonate spin-label (MTSSL), have been studied using DQC-ESR at 9 and 17 GHz. The distances span the range from 20 Å for the 65∕76 mutant to 47 Å for the 61∕135 mutant. The high quality of the dipolar spectra also allows the determination of the distance distributions, the width of which can be used to set upper and lower bounds in future computational strategy. It is also demonstrated that the shape of these distributions can reveal the presence of multiple conformations of the spin-label, an issue of critical relevance to the structural interpretation of the distances. The distances and distributions found in this study are readily rationalized in terms of the known crystal structure, the characteristic conformers of the nitroxide side chains, and molecular modeling. This study sets the stage for the use of DQC-ESR for determining the tertiary structure of large proteins with just a small number of long-distance constraints.
|
|
|
A Novel View of Domain Flexibility in E. coli Adenylate Kinase Based on Structural Mode-Coupling 15N NMR Relaxation
V. Tugarinov, Y.E. Shapiro, Z. Liang, J.H. Freed and E. Meirovitch.
J. Mol. Biol. 315, 155-170 (2002).
Supporting Information
<doi: 10.1006/jmbi.2001.5231>
PMID:
11779236
|
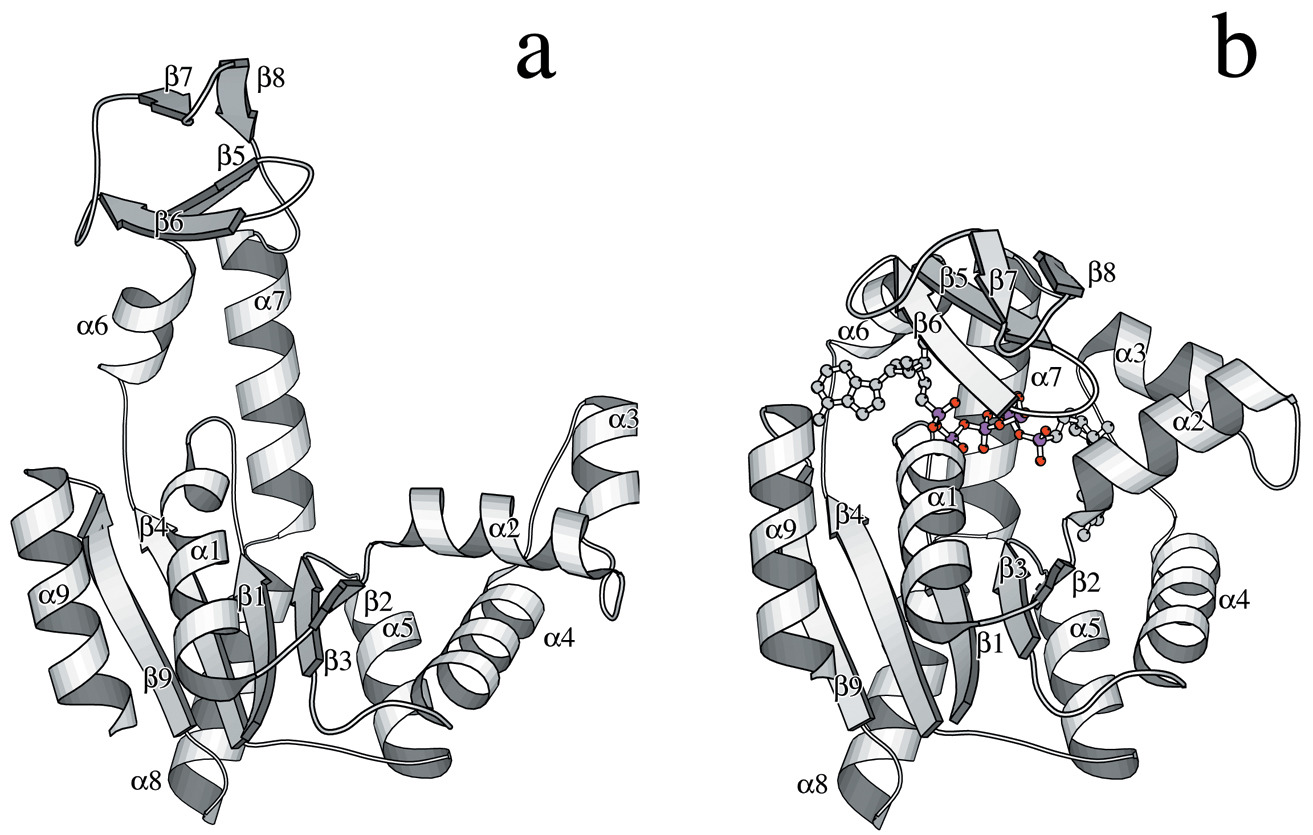
|
|
ABSTRACT: Adenylate kinase from Escherichia coli (AKeco), consisting of a single 23.6 kDa polypeptide chain folded into domains CORE, AMPbd and LID, catalyzes the reaction AMP+ATP→2ADP. In the ligand-free enzyme the domains AMPbd and LID execute large-amplitude movements controlling substrate binding and product release during catalysis. Domain flexibility is investigated herein with the slowly relaxing local structure (SRLS) model for 15N relaxation. SRLS accounts rigorously for coupling between the global and local N-H motions through a local ordering potential exerted by the protein structure at the N-H bond. The latter reorients with respect to its protein surroundings, which reorient on the slower time scale associated with the global protein tumbling. AKeco diffuses globally with correlation time τm=15.1 ns, while locally two different dynamic cases prevail. The domain CORE features ordering about the equilibrium N-H bond orientation with order parameters, S2, of 0.8-0.9 and local motional correlation times, τ, mainly between 5-130 ps. This represents a conventional rigid protein structure with rapid small-amplitude N-H fluctuations. The domains AMPbd and LID feature small parallel (ZM) ordering of S2=0.2-0.5 which can be reinterpreted as high perpendicular (YM) ordering. M denotes the local ordering/local diffusion frame. Local motion about ZM is given by τ∥≈5 ps and local motion of the effective ZM axis about YM by τ⊥=6-11 ns. ZM is tilted at approximately 20° from the N-H bond. The orientation of the YM axis may be considered parallel to the Ci−1α-Ciα axis. The τ⊥ mode reflects collective nanosecond peptide-plane motions, interpretable as domain motion. A powerful new model of protein flexibility∕domain motion has been established. Conformational exchange (Rex) processes accompany the τ⊥ mode. The SRLS analysis is compared with the conventional model-free analysis.
|
|
|
Single-Crystal EPR Studies of Transition-Metal Ions in Inorganic Crystals at Very High Frequency
S.K. Misra, S.I. Andronenko, K.A. Earle, J.H. Freed.
Appl. Magn. Reson. 21, 549-561 (2001).
<doi: 10.1007/BF03162428>
|
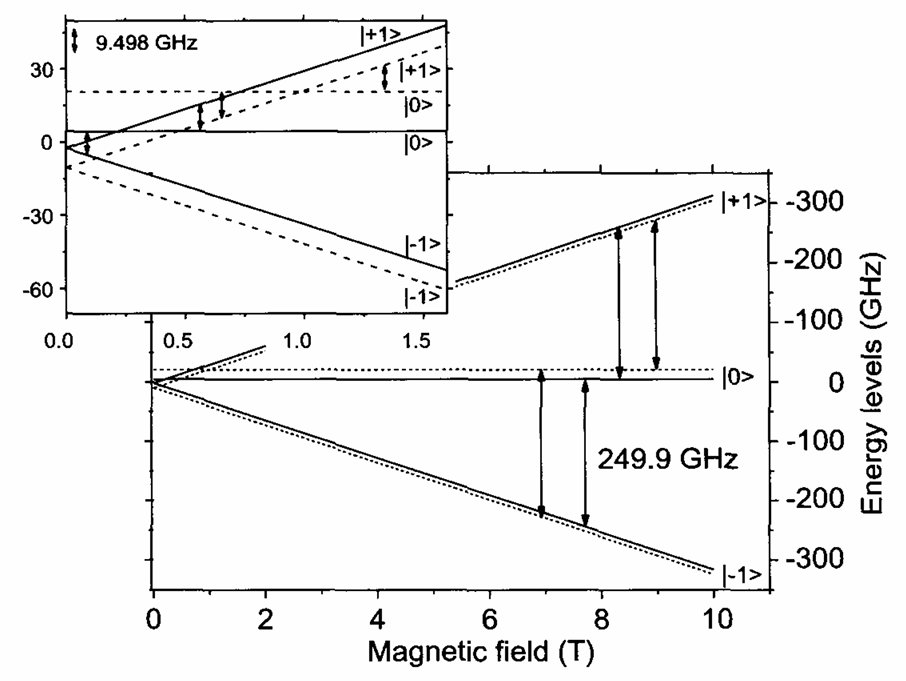
|
|
ABSTRACT: Electron paramagnetic resonance (EPR) single-crystal rotation studies at very high frequency (249.9 GHz) of transition metal ions with electron spins greater than one-half are reported. At 249.9 GHz, the spectra are in the high-field limit despite large zero-field splittings. This leads to a considerable simplification of the spectra, and aids in their interpretation. Single-crystal 249.9 GHz EPR spectra of Ni2+ in Ni2CdCl6 ⋅ 12H2O, Mn2+ (0.2%) in ZnV2O7, and Fe3+ (2%) in CaYAl04 were recorded at 253 K in an external magnetic field of up to 9.2 T, along with those at X-band and Q-band frequencies at 295 K and lower temperatures. The goniometer used at 249.9 GHz for single-crystal rotation is based on a quasi-optical design and is an integral part of a special Fabry-Pérot resonator. The values of the spin-Hamiltonian parameters were estimated from a simultaneous fitting of all of the observed line positions at several microwave frequencies recorded at various orientations of each crystal with respect to the external magnetic field with least-squares fitting in conjunction with matrix diagonalization. Estimates of zero-field splitting parameter D at room temperature are: for Ni2+, about −31 GHz (site I) and about −7 GHz (site II); for Mn2+, about 6 GHz; and for Fe3+, about 29 GHz.
|
|
|
A Multifrequency ESR Study of the Complex Dynamics of Membranes
Y. Lou, M. Ge, and J.H. Freed.
J. Phys. Chem. B 105, 11053-11056 (2001).
<doi: 10.1021/jp013226c>
|
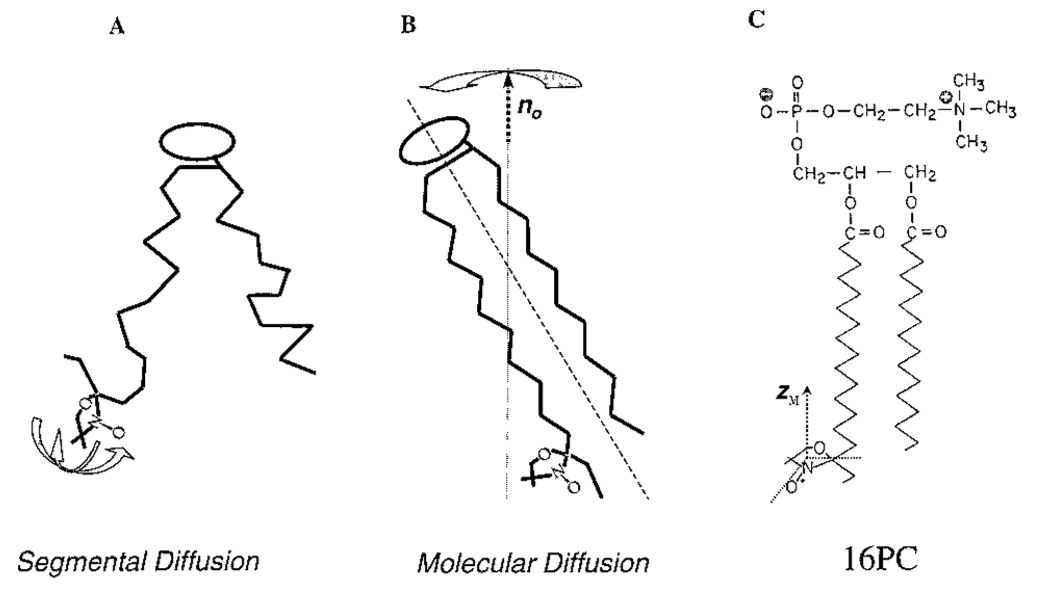
|
|
ABSTRACT: A combined 250 and 9 GHz ESR study was performed on membrane vesicles composed of pure lipid (DPPC) and of DPPC:cholesterol in a 1:1 molar ratio using the end chain labeled lipid, 16-PC. It is shown that the 250 GHz spectra represent a "fast time-scale" such that the overall restricted motion of the lipid in the membrane is frozen out, but it is sensitive to the internal dynamics of the end chain. The 9 GHz spectra are, however, sensitive to the overall motion as well. This combined study thus permitted the separation of both types of motion. It is shown that the addition of cholesterol has a large effect on the end chain dynamics by restricting its motion while increasing motional rates, whereas its effects on the overall lipid motion are modest. This leads to a clearer characterization of the dynamic structure of the cholesterol-rich liquid-ordered phase as compared to the liquid crystalline phase. This study thus shows the significant potential of a multifrequency ESR approach to the complex dynamics of membranes.
|
|
|
ADP Ribosylation Factor 6 Binding to Phosphatidylinositol 4,5-Bisphosphate-Containing Vesicles Creates Defects in the Bilayer Structure: An Electron Spin Resonance Study
M. Ge, J.S. Cohen, H.A. Brown, and J.H. Freed.
Biophys. J. 81, 994-1005 (2001).
<doi: 10.1016/S0006-3495(01)75757-8>
PMID:
11463641
PMCID:
PMC1301569
|
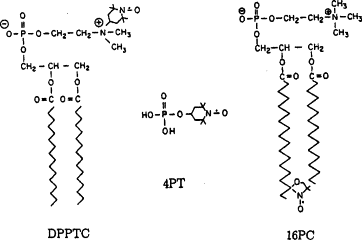
|
|
ABSTRACT: The effects of binding of myristoylated ADP ribosylation factor 6 (myr-ARF6), an activator of phospholipase D (PLD), to a model membrane were investigated using an electron spin resonance (ESR) labeling technique. Initial studies were conducted in vesicles composed of 1-palmitoyl-2-oleoyl phosphatidylethanolamine, dipalmitoylphosphatidylcholine, phosphatidylinositol 4,5-biphosphate (PIP2), and cholesterol. Recombinant ARF6 binding significantly enhances defects in both the headgroup and acyl-chain regions of the membrane, which are revealed by the emergence of sharp components in the spectra from a headgroup label, 1,2-dipalmitoylphosphatidyl-2,2,6,6-tetramethyl-1-piperidinyloxy-choline (DPPTC), and a chain label, 10PC, after myr-ARF6 binding. Binding of non-myristoylated ARF6 (non-ARF6) shows markedly reduced effects. Interestingly, no change in spectra from DPPTC was observed upon myr-ARF6 binding when PIP2 in the vesicles was replaced by other negatively charged lipids, including phosphatidylinositol, phosphatidylserine, and phosphatidylglycerol, even when normalized for charge. The production of the sharp peak appears to be a specific event, because another GTP binding protein, CDC42, which binds PIP2 and activates PLD, fails to induce changes in vesicle structure. These results suggest a previously unappreciated role for ARF in mediating a protein/lipid interaction that produces defects in lipid bilayers. This function may serve as an initial event in destabilizing membrane structure for subsequent membrane fusion or biogenesis of vesicles.
|
|
|
A Many-Body Analysis of the Effects of the Matrix Protons and their Diffusional Motion on Electron Spin Resonance Line Shapes and Electron Spin Echoes
A.A. Nevzorov and J.H. Freed.
J. Chem. Phys. 115, 2416-2429 (2001)
<doi: 10.1063/1.1382817>
|
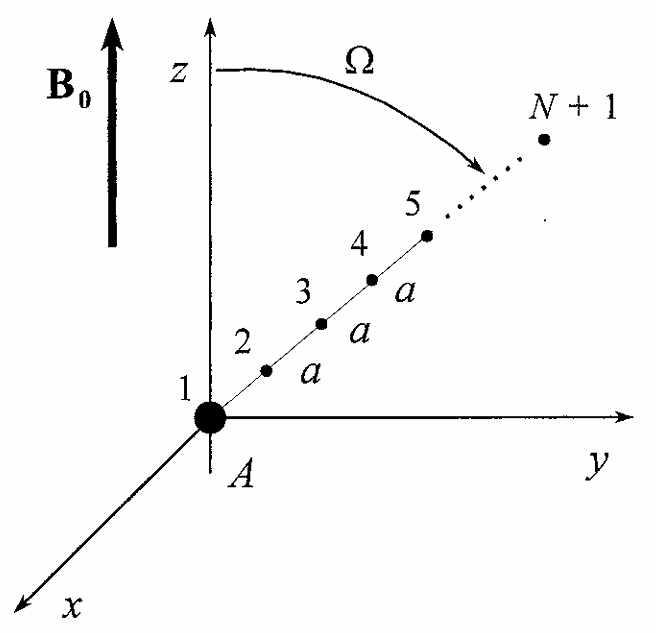
|
|
ABSTRACT: The method for treating the evolution of the density matrix developed in the accompanying paper for many-spin systems is applied here for calculating magnetic resonance signals of a spin A interacting with a bath of N identical spins B. Spins B are assumed to have much smaller gyromagnetic ratios than the spin A (e.g., the former are nuclear spins, I and the latter is an electron spin, S). The experimentally observed quadratic dependence of the spin-echo envelope decay on concentration and time is explained from considering the dipolar coupling of spin A to all the B spins in the presence of B–B dipolar interactions. It is shown that the spin-echo envelope decay in the rigid limit is due to the interaction of the A spin with the coherent many-body states of the coupled spins B via the nuclear flip-flop terms I±I∓ which becomes a dissipative mechanism in the thermodynamic limit. This represents a more rigorous analysis than simplified models based on an incoherent version of "spin diffusion," and it leads to good quantitative agreement with experiment. Moreover, this analysis represents a unified description of both the modulation and decay of the A-spin echoes. Spin echoes and line shapes for the A–BN systems are also calculated for finite motions which randomize the B spins. Even for very slow motions (modeled as translational diffusion) an effective mechanism for spin-echo envelope decay is generated, which readily overtakes the coherent mechanism in importance. The intensity distribution for the forbidden components in the A-spin line shape resulting from multiquantum transitions of the B spins caused by the pseudosecular interaction terms SzI± is calculated. In the rigid limit it is found to behave like a Poisson distribution.
|
|
|
Direct-Product Formalism for Calculating Magnetic Resonance Signals in Many-Body Systems of Interacting Spins
A.A. Nevzorov and J.H. Freed.
J. Chem. Phys. 115, 2401-2415 (2001).
<doi: 10.1063/1.1382816>
|
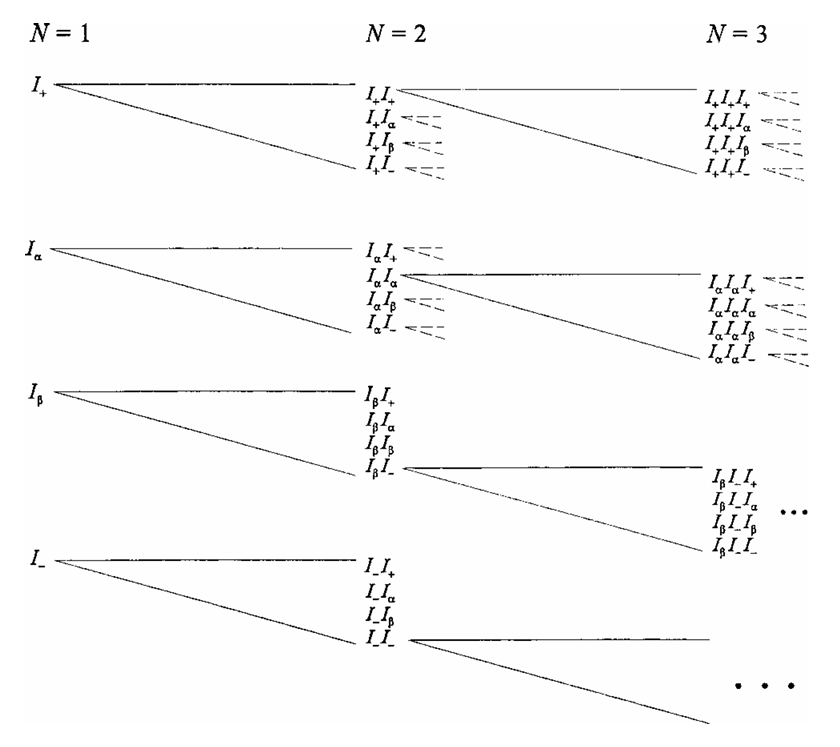
|
|
ABSTRACT: Modern applications of nuclear magnetic resonance and electron spin resonance, and especially quantum computing problems call for more effective formalisms to describe relaxation and evolution of various orders of coherence in the presence of motions of the interacting spins. Here we suggest a formulation for the description of multiquantum spin states based on direct-product structures that take into account the inherent permutation symmetries and quantum coherences of a multispin system. Convenient recursion relations are obtained for the matrix representations of the N-body Hamiltonian superoperator and pulse propagators. This allows one to obtain compact expressions for the evolution of magnetic resonance signals when the dipolar interactions amongst spin-bearing molecules are modulated by their motions. These expressions include the free-induction decay and solid echoes, as well as the decay of higher-order coherences under the assumption of statistical independence of the motions of the spin-bearing molecules. Exact results, not requiring this assumption, are obtained for the case of the truncated dipolar Hamiltonian. Important phenomena that arise in multispin systems, such as instantaneous diffusion and spectral diffusion arising from motions, are studied more rigorously by solving the equation for the time evolution of the spin-density states. The many-body magnetic-resonance signals in the presence of motions are obtained by solving the appropriate stochastic Liouville equations. These solutions may be compared to solid-echo experiments to extract translational diffusion coefficients even in the slow motional regime.
|
|
|
A Structural Mode-Coupling Approach to 15N NMR Relaxation in Proteins
V. Tugarinov, Z. Liang, Y.E. Shapiro, J.H. Freed, and E. Meirovitch.
J. Am. Chem. Soc. 123, 3055-3063 (2001).
Supporting Information
<doi: 10.1021/ja003803v>
PMID:
11457016
|
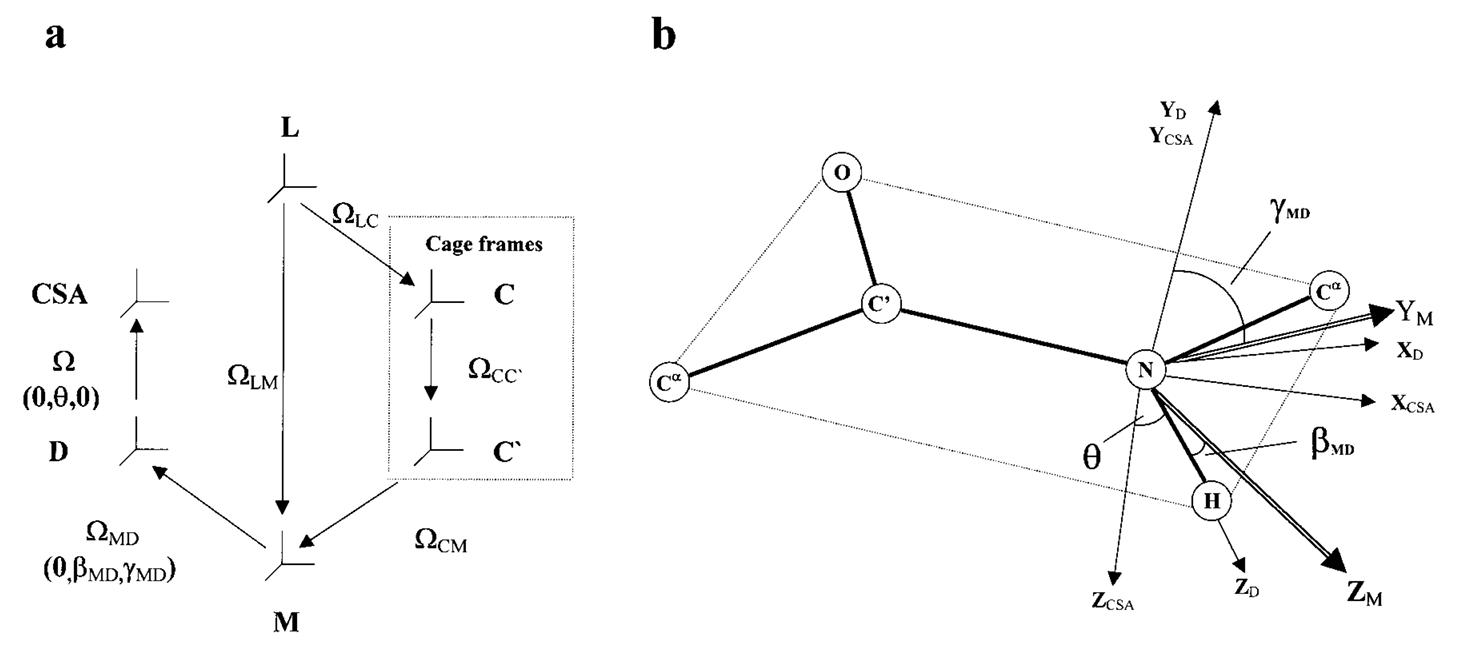
|
|
ABSTRACT: The two-body Slowly Relaxing Local Structure (SRLS) model was applied to 15N NMR spin relaxation in proteins and compared with the commonly used original and extended model-free (MF) approaches. In MF, the dynamic modes are assumed to be decoupled, local ordering at the N−H sites is represented by generalized order parameters, and internal motions are described by effective correlation times. SRLS accounts for dynamical coupling between the global diffusion of the protein and the internal motion of the N−H bond vector. The local ordering associated with the coupling potential and the internal N−H diffusion are tensors with orientations that may be tilted relative to the global diffusion and magnetic frames. SRLS generates spectral density functions that differ from the MF formulas. The MF spectral densities can be regarded as limiting cases of the SRLS spectral density. SRLS-based model-fitting and model-selection schemes similar to the currently used MF-based ones were devised, and a correspondence between analogous SRLS and model-free parameters was established. It was found that experimental NMR data are sensitive to the presence of mixed modes. Our results showed that MF can significantly overestimate order parameters and underestimate local motion correlation times in proteins. The extent of these digressions in the derived microdynamic parameters is estimated in the various parameter ranges, and correlated with the time scale separation between local and global motions. The SRLS-based analysis was tested extensively on 15N relaxation data from several isotropically tumbling proteins. The results of SRLS-based fitting are illustrated with RNase H from E. coli, a protein extensively studied previously with MF.
|
|
|
Electron Spin Resonance in Studies of Membranes and Proteins
P.P. Borbat, A.J. Costa-Filho, K.A. Earle, J.K. Moscicki, J.H. Freed.
Science 291, 266-269, (2001).
<doi: 10.1126/science.291.5502.266>
PMID:
11253218
|
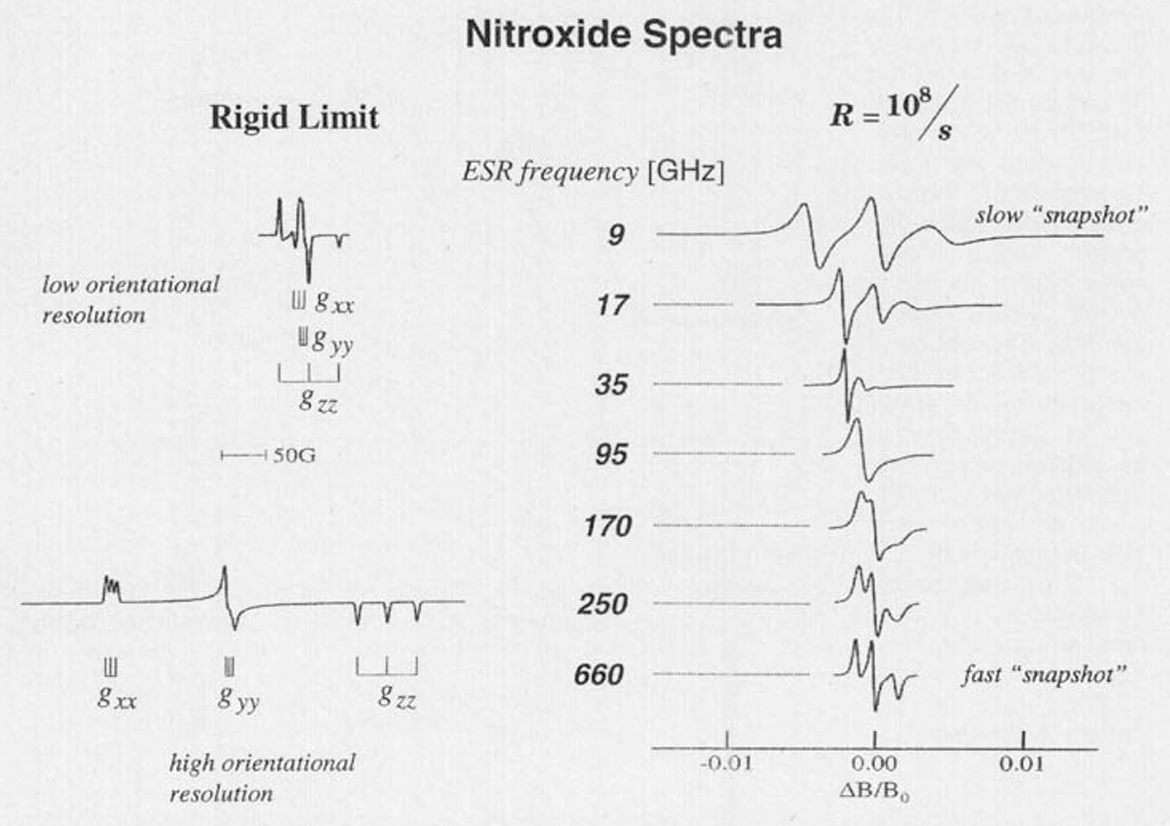
|
|
ABSTRACT: We provide a review of current electron spin resonance (ESR) techniques for studying basic molecular mechanisms in membranes and proteins by using nitroxide spin labels. In particular, nitroxide spin label studies with high-field/high-frequency ESR and two-dimensional Fourier transform ESR enable one to accurately determine distances in biomolecules, unravel the details of the complex dynamics in proteins, characterize the dynamic structure of membrane domains, and discriminate between bulk lipids and boundary lipids that coat transmembrane peptides or proteins; these studies can also provide time resolution to studies of functional dynamics of proteins. We illustrate these capabilities with recent examples.
|
|
|
New Technologies in Electron Spin Resonance
J.H. Freed.
Annu. Rev. Phys. Chem. 51, 655-689 (2000).
<doi: 10.1146/annurev.physchem.51.1.655>
PMID:
11031296
|
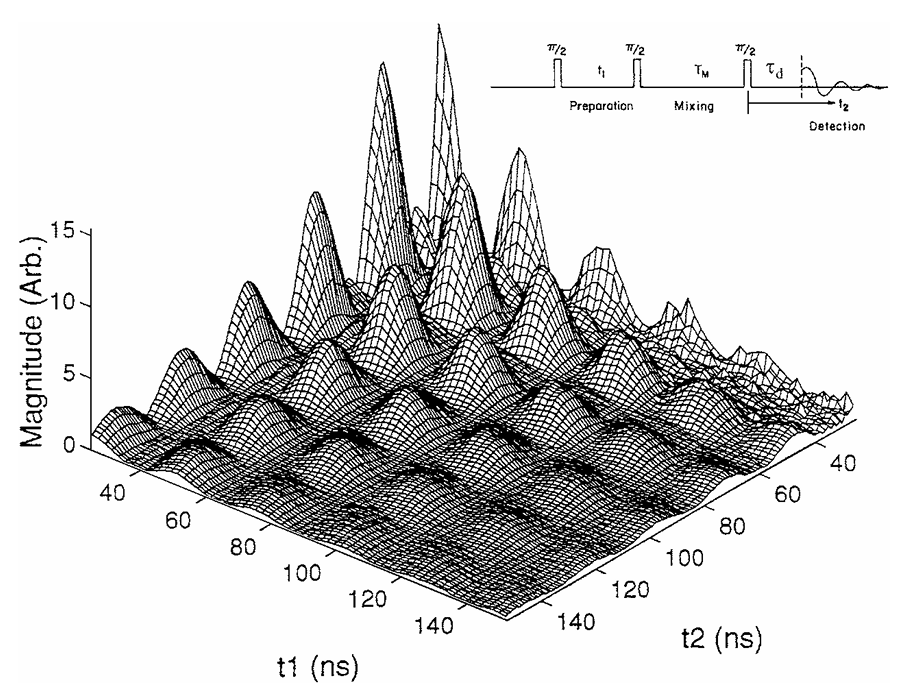
|
|
ABSTRACT: New electron spin resonance (ESR) technologies have been developed, which have led to new and improved applications. (a) The development of two-dimensional Fourier transform (FT) ESR required spectrometers providing intense π∕2 microwave pulses of very short (3–5 ns) duration, wide bandwidths, and very short dead times. It has enabled studies that resolve sophisticated details of molecular dynamics in complex fluids. (b) Methods that produce multiple quantum coherences by pulsed ESR now enable accurate measurements of large distances (>12Å). (c) One of the most important advances has been the extension of ESR to high magnetic fields and high frequencies. This has benefited from the utilization of quasi-optical methods, especially above 150 GHz. The greatly improved orientational resolution and the faster "snapshot" of motions that are provided by ESR at high frequencies enhance studies of molecular dynamics. The use of both high and lower frequencies enables one to unravel faster and slower modes from the complex dynamics of fluids and macromolecules. (d) The development of FT-ESR imaging required substantial pulsed field gradients lasting only 50–100 ns. ESR imaging is effective in studying diffusion in fluids. Areas for further development are also described.
|
|
|
Double Quantum ESR and Distance Measurements
P.P. Borbat and J.H. Freed.
In Distance Measurements in Biological Systems by EPR, L.J. Berliner, S.S. Eaton, and G.R, Eaton, Eds.
Biological Magnetic Resonance 19, Chapter 9, pp. 383-459 (2000). (Kluwer, NY).
|
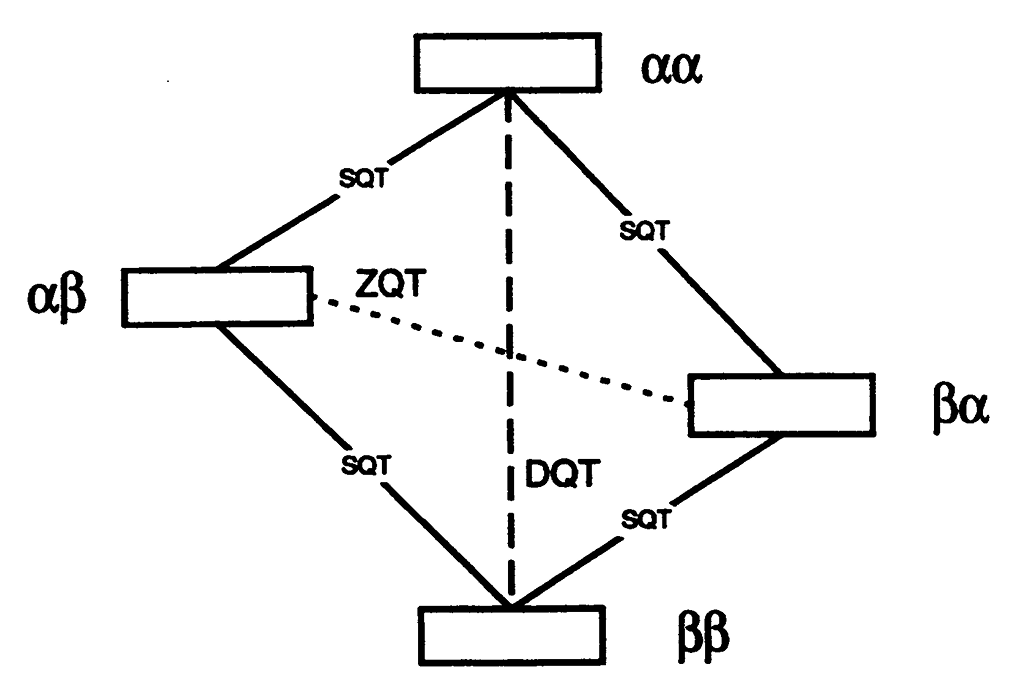
|
|
Double Quantum ESR and Distance Measurements
P.P. Borbat and J.H. Freed.
In Distance Measurements in Biological Systems by EPR, L.J. Berliner, S.S. Eaton, and G.R, Eaton, Eds.
Biological Magnetic Resonance 19, Chapter 9, pp. 383-459 (2000). (Kluwer, NY).
<doi: 10.1007/0-306-47109-4_9>
|
|
|
ABSTRACT: "Allowed" double quantum coherences (DQC) can now be routinely generated in disordered and oriented solids containing nitroxide biradicals and random distributions of stable radicals. The Pake doublets obtained from DQC pathways can be effectively used to determine a broad range of distances in the former case whereas decay constants yield concentrations in the latter. The DQC signals are strong and often comparable to standard single quantum signals. They are free of any large undesirable signals, so the DQ experiment is easy to perform. Their strong intensity permits the study of low concentrations of spins in samples typical of those ordinarily met in the case of doubly-labeled macromolecules such as proteins and polypeptides. The upper range of distances for systems labeled with nitroxides is estimated to be ca. 80 Å. In the limit of non-selective pulses the interpretation of DQC signals becomes independent of complicating geometric features which affect other ESR distance methods. The method is compared to other existing pulse distance measurement techniques and future improvements are also discussed.
|
|
|
Segmental Rotational Diffusion of Spin-Labeled Polystyrene in Dilute Toluene Solution by 9 and 250 GHz ESR
J. Pilar, J. Labský, A. Marek, D.E. Budil, K.A. Earle, and J.H. Freed.
Macromolecules 33, 4438-4444 (2000).
<doi: 10.1021/ma0002242>
|
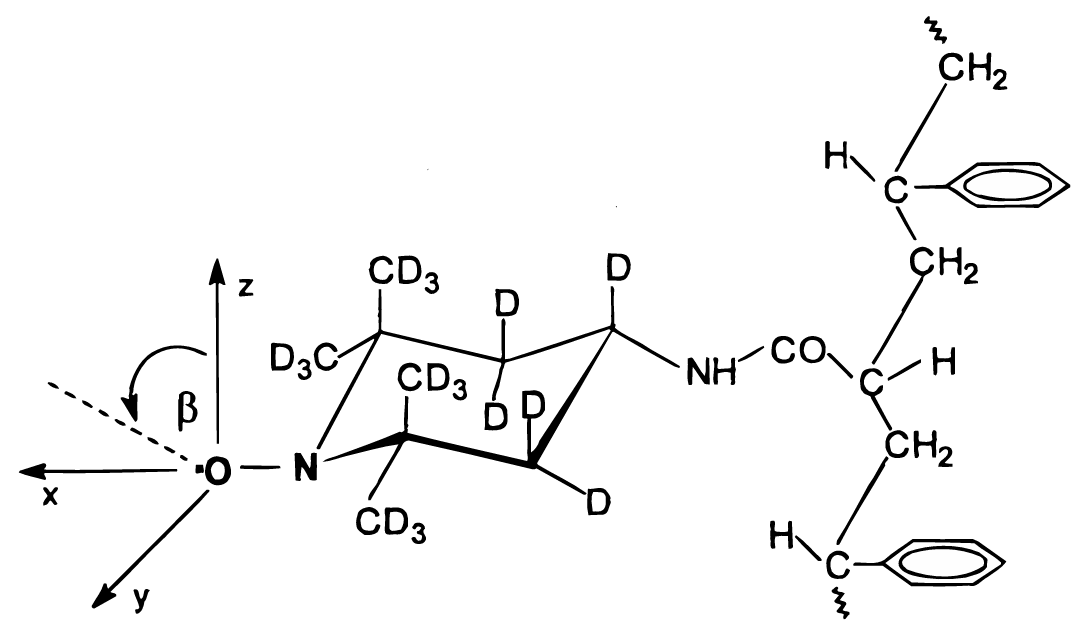
|
|
ABSTRACT: A copolymer of styrene containing less than 5 mol % of chain units spin-labeled by attachment of the nitroxide via a short tether has been synthesized. ESR spectra of dilute toluene solution of the copolymer have been obtained using 9 and 250 GHz ESR. Parameters characterizing polystyrene segmental rotational diffusion in toluene solution over a broad temperature range have been determined from nonlinear least-squares fits of theoretical ESR spectra to the experimental ESR spectra. The model used was that of relatively fast internal rotation of the nitroxide about its tether, with slower polymer chain segmental motion. Together they lead to effective rotational diffusion with an anisotropic diffusion tensor. In addition, constraints in the form of an orientational potential restrict the range of angles over which this diffusion occurs relative to the polymer backbone, and the latter is assumed to reorient on an ultraslow time scale. This is referred to as a model of microscopic order but macroscopic disorder (MOMD). Rates for the slower polymer chain segmental motion (from the 250 GHz spectra) ranged from 3.6 × 108 s-1 at 311 K to 0.15 × 108 s-1 at 215 K, with a substantial orientational potential of about 2kT over this temperature range. Although there was reasonable agreement between the results obtained at 9 and 250 GHz, there were systematic discrepancies such that the orienting potentials obtained from the 250 GHz spectra were about twice (or more) those from the 9 GHz spectra, and the rotational diffusion tensor components from the 250 GHz spectra were at least twice those from the 9 GHz spectra. This implies a breakdown of the MOMD model for the 9 GHz spectra presumably due to their sensitivity to the slower overall tumbling motion at this low spectral frequency. For the faster "time scale" of the 250 GHz spectra, such a slow motion is "frozen out", rendering these spectra consistent with the MOMD model. Nevertheless, the results at both frequencies yielded a common activation energy, Eexp = 20.7 ± 1.5 kJ∕mol, which, when corrected for the viscous flow contribution, yielded an Ea = 11.9 ± 1.5 kJ∕mol, which is in good agreement with recent results from fluorescence studies.
|
|
|
An Electron Spin Resonance Study of DNA Dynamics Using the Slowly Relaxing Local Structure Model
Z. Liang, J.H. Freed, R.S. Keyes and A.M. Bobst.
J. Phys. Chem. B 104, 5372-5381 (2000).
<doi: 10.1021/jp994219f>
|
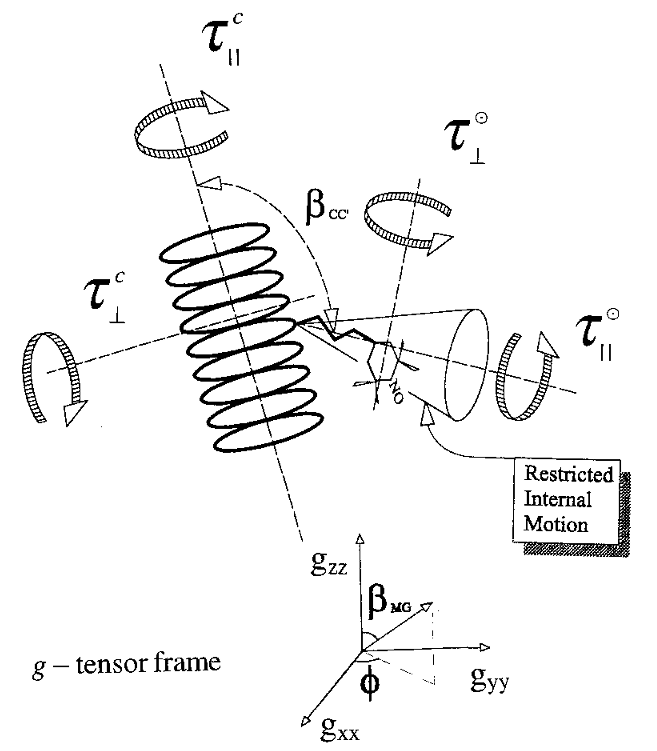
|
|
ABSTRACT: A systematic analysis is presented of the ESR spectra from DNA oligomers of a range of sizes, including a polymer, spin-labeled with nitroxide moieties attached by different tethers. The complexity of the DNA dynamics is dealt with by the use of the general slowly relaxing local structure (SRLS) model, wherein the nitroxide moiety is reorienting in a restricted local environment, which itself is relaxing on a longer time scale. The slower motion describes the global tumbling of the DNA lattice, and the faster motion, the internal dynamics. In the present analysis, the correlation times for the axially symmetric global tumbling were those obtained from hydrodynamic theory, while the correlation times for the internal dynamics and the order parameter, which directly measures its restricted range of motion, were determined by nonlinear least-squares fits to the spectra. The principal result is the observation and characterization of two types of spectra from these labeled DNA systems. These two spectra represent components that differ from each other with respect to their local environments, one a highly restricted site yielding a large order parameter (0.61) and slower internal motions and the other a much less restricted site (with order parameter of 0.18) and faster internal motions. Whereas the one-atom tethered DUTA exhibits both sites (in roughly 9:1 ratio with the more restricted site more prevalent), the two-atom tethered DUMTA and five-atom tethered DUAT exhibit just the more restricted site, but the five-atom tethered DUAP exhibits only the less restricted site. (DUAP differs from DUAT and the others in having a less flexible tether.) It is suggested that the spin labels trapped in the highly restricted∕slow motional site have a stronger interaction with the base than those in the other site. In general, the longer the tether, the faster are the correlation times for internal dynamics. For all tethers, it is found that the correlation time for internal motion perpendicular to the internal symmetry axis systematically becomes slower as the size of the oligomer increases. It is suggested that this may be a manifestation of collective modes of motion of the DNA. It is pointed out that the simpler models used in previous ESR studies are simplified cases of the more realistic SRLS model.
|
|
|
Dipolar Relaxation in a Many-Body System of Spins of 1/2
A.A. Nevzorov and J.H. Freed.
J. Chem. Phys. 112, 1425-1443 (2000).
<doi: 10.1063/1.480709>
|
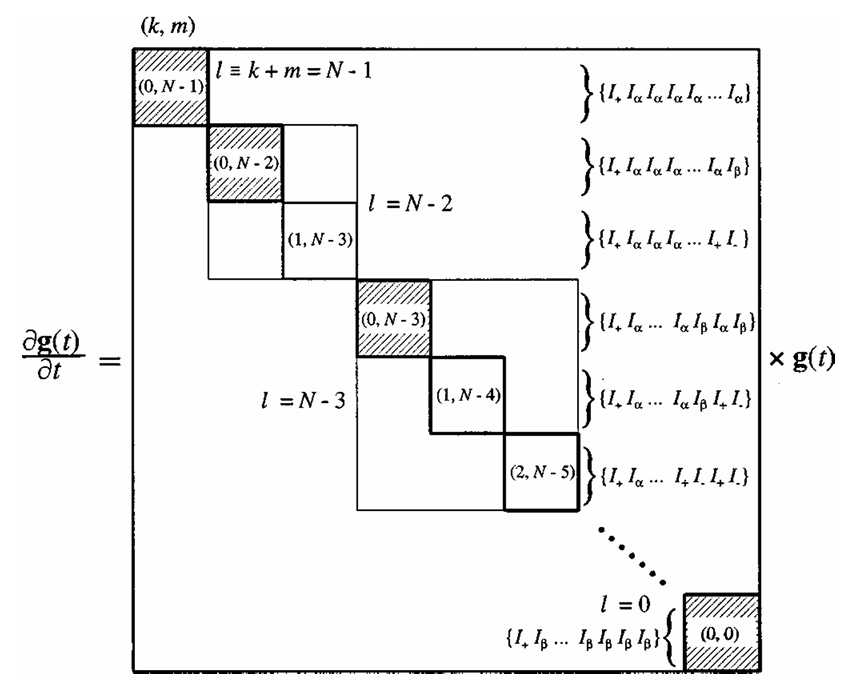
|
|
ABSTRACT: The method utilized in Paper I [J. Chem. Phys. 112, 1413 (2000)] for treating the density matrix equation for a two-spin system in the presence of the dipolar interaction that is randomly modulated by translational diffusion, is extended to a many-body system of identical spins of 1∕2. Generalized cumulant expansions are used, which allow one to take full advantage of the statistical independence of the motions of spins. In the high-temperature approximation (appropriate for dilute solutions), for a single nonselective pulse, the symmetry of the problem allows one to obtain a compact ordered binomial expression for the free-induction decay signal that is related to the two-particle solution, and it still contains the two spin-isochromat components. The latter are evaluated by solving the corresponding stochastic Liouville equation, which allows one to recover in a unified way the two limiting cases including Anderson's result for statistical broadening in a rigid lattice and the classical Torrey–Bloembergen–Redfield expression for the motional narrowing, as corrected by Hwang and Freed. The line shape expression in the thermodynamic limit, i.e., for large numbers of particles in a macroscopic volume, is obtained. It is found that the many-body dipolar line shapes are very close to Lorentzians over the entire motional range studied, with the linewidths proportional to the spin concentration, as predicted earlier for the limiting cases. Linewidths plotted versus the values of the translational diffusion coefficient clearly show the solid-state limit, the motional-narrowing limit, and the intermediate region. The method is extended to describe the behavior of the many-body system in a solid-echo sequence. This enables one to obtain the homogeneous T2's over the whole range of motions. A minimum in T2 is found at approximately the same value of translational diffusion coefficient as was found for the two-spin case in Paper I.
|
|
|
Spin Relaxation by Dipolar Coupling: From Motional Narrowing to the Rigid Limit
A.A. Nevzorov and J.H. Freed.
J. Chem. Phys. 112, 1413-1424 (2000).
<doi: 10.1063/1.480695>
|
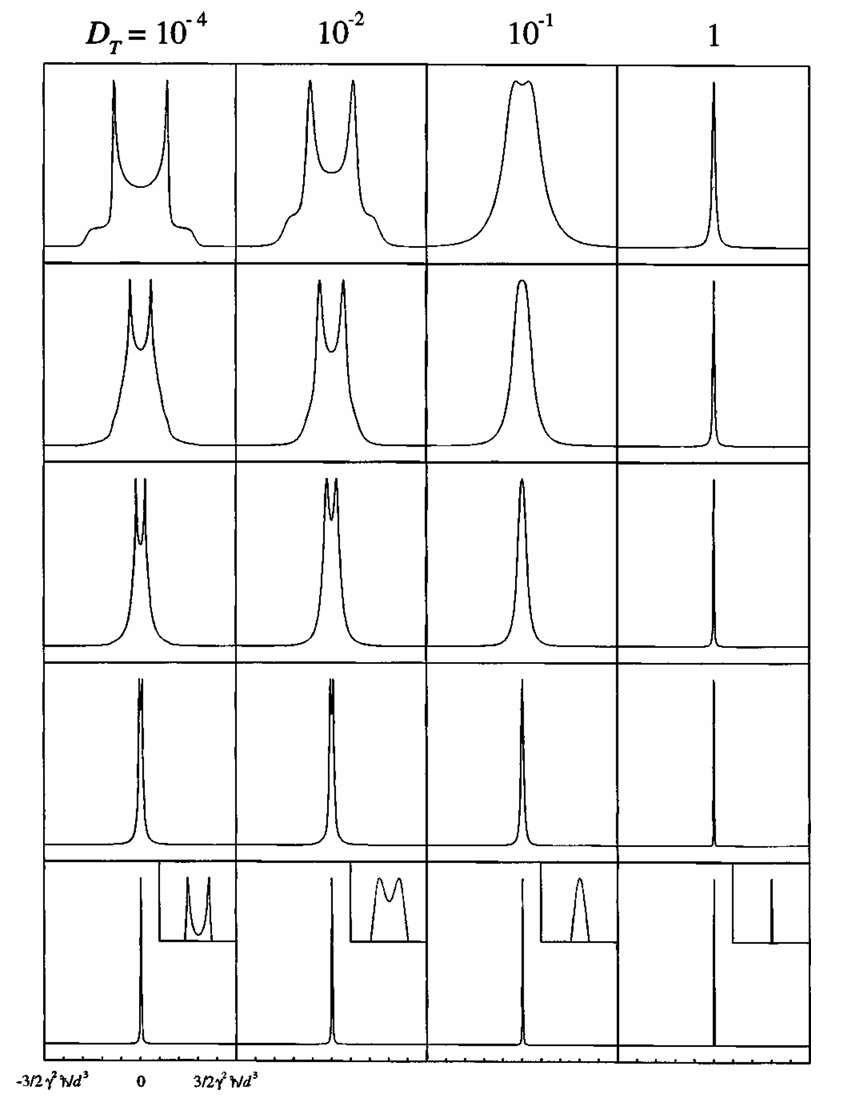
|
|
ABSTRACT: A coupled system of two molecules bearing spins of 1∕2, which are allowed to diffuse relative to each other, is considered. By using a symmetry-adapted basis operator set, the overall density matrix equation is decoupled into two equations for the time-resolved isochromat components, the sum of which yields the observed signal. The appropriate stochastic Liouville equation is solved by a combination of eigenfunction expansion and finite-differences for the angular and radial relative motions, respectively. A full range of spectra from classic Pake patterns in the rigid limit to motionally narrowed Lorentzians is recovered. As an extension of the above approach, the solid-echo experiment is described in terms of the ensemble-averaged isochromats. Homogeneous transverse relaxation times (T2) as a function of the translational diffusion coefficient (DT) are obtained from simulating SECSY (spin-echo correlation spectroscopy) signals, which show distinct T2 minima vs DT. The present method of separating the quantum and stochastic classical variables constitutes a useful approach for treating multiquantum statistical systems, and it can be generalized to an arbitrary number of spins as shown in a companion paper. In the present study we obtained the usual linear dependence of T2 on DT in the motional narrowing (or Redfield) limit, whereas in the slow motional regime a DT−1∕2 dependence is observed, consistent with studies of rotational diffusion. Varying the distance of maximum separation between the two spins (rmax) has virtually no effect on the location of the T2 minimum with respect to DT, implying that the onset of slow motion is essentially independent of rmax.
|
|
|
Multiple-Quantum ESR and Distance Measurements
P.P. Borbat and J.H. Freed.
Chem. Phys. Lett. 313, 145-154 (1999).
<doi: 10.1016/S0009-2614(99)00972-0>
|
|
|
ABSTRACT: It is shown that allowed double-quantum coherences (DQC) can now be routinely generated in disordered and oriented solids containing nitroxide biradicals and random distributions of stable radicals. The Pake doublets obtained from DQC pathways can be effectively used to determine long (˜30 Å) distances in the former case, and concentrations in the latter. The DQC signals are strong and often comparable to standard single-quantum signals. In the limit of non-selective pulses their interpretation becomes independent of complicating features which affect other ESR distance methods.
|
|
|
Electron Spin Resonance Characterization of Liquid Ordered Phase of Detergent Resistant Membranes from RBL-2H3 Cells
M. Ge, K.A. Field, R. Aneja, D. Holowka, B. Baird, and J.H. Freed.
Biophys. J. 77, 925-933 (1999).
<doi: 10.1016/S0006-3495(99)76943-2>
PMID:
10423437
PMCID:
PMC1300383
|

|
|
ABSTRACT: The dynamic structure of detergent-resistant membranes (DRMs) isolated from RBL-2H3 cells was characterized using two different acyl chain spin-labeled phospholipids (5PC and 16PC), a headgroup labeled sphingomyelin (SM) analog (SD-Tempo) and a spin-labeled cholestane (CSL). It was shown, by comparison to dispersions of SM, dipalmitoylphosphatidylcholine (DPPC), and DPPC∕cholesterol of molar ratio 1, that DRM contains a substantial amount of liquid ordered phase: 1) The rotational diffusion rates (R⊥) of 16PC in DRM between −5°C and 45°C are nearly the same as those in molar ratio DPPC∕Chol=1 dispersions, and they are substantially greater than R⊥ in pure DPPC dispersions in the gel phase studied above 20°C; 2) The order parameters (S) of 16PC in DRM at temperatures above 4°C are comparable to those in DPPC∕Chol=1 dispersions, but are greater than those in DPPC dispersions in both the gel and liquid crystalline phases. 3) Similarly, R⊥ for 5PC and CSL in DRM is greater than in pure SM dispersions in the gel phase, and S for these labels in DRM is greater than in the SM dispersions in both the gel and liquid crystalline phases. 4) R⊥ of SD-Tempo in DRM is greater than in dispersions of SM in both gel and liquid phases, consistent with the liquid-like mobility in the acyl chain region in DRM. However, S of SD-Tempo in DRM is substantially less than that of this spin label in SM in gel and liquid crystalline phases (in absolute values), indicating that the headgroup region in DRMs is less ordered than in pure SM. These results support the hypothesis that plasma membranes contain DRM domains with a liquid ordered phase that may coexist with a liquid crystalline phase. There also appears to be a coexisting region in DRMs in which the chain labels 16PC and 5PC are found to cluster. We suggest that other biological membranes containing high concentrations of cholesterol also contain a liquid ordered phase.
|
|
|
An Assessment of the Applicability of Multifrequency ESR to Study the Complex Dynamics of Biomolecules
Z. Liang and J.H. Freed.
J. Phys. Chem. B 103, 6384-6396 (1999).
<doi: 10.1021/jp9907746>
|
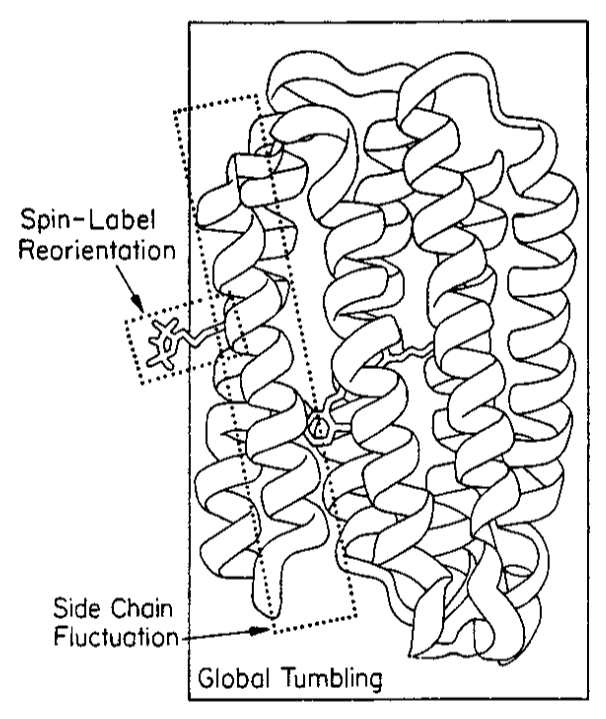
|
|
ABSTRACT: It is shown that the commonly used models for analyzing ESR spectra from nitroxide spin-labeled proteins or DNA systems are special cases of the more general slowly relaxing local structure (SRLS) model, wherein the nitroxide spin probe is taken as reorienting in a restricted local environment, which itself is relaxing on a longer time scale. This faster motion describes the internal dynamics, while the slower motion describes the global tumbling of the macromolecule. By using the SRLS model as the reference, it is shown (1) under what conditions the microscopic-order macroscopic-disorder (MOMD) model, wherein the global tumbling of the macromolecule is in the rigid limit, is valid, and (2) when the fast internal motion (FIM) model, wherein the internal motion is so rapid as to lead to partial averaging of the magnetic tensors, is valid. The frequency dependence of these models is studied. A key general property of high frequency ESR that emerges is that it reports on a faster motional time scale, whereas low frequency ESR reports on a slower motional time scale. It is shown that, in general, the MOMD model is a better approximation for ESR spectra obtained at high frequency (250 GHz), whereas, in general, the FIM model is a better approximation for low frequency (9 GHz) ESR spectra. However, in general, one does not find that the simpler model fits, at a single ESR frequency, to the more complete SRLS model, return correct motional and ordering parameters. The simultaneous fitting of both low and high frequency ESR spectra is thus required to remove such ambiguities and to return all the various dynamic, ordering, and geometric factors that characterize the complex dynamics. This approach is briefly related to recent ESR spectra from the spin-labeled protein, T4 lysozyme, and from spin-labeled DNA nucleosides. In order to better apply the slow-motional SRLS model to macromolecular dynamics, the Polimeno−Freed theory has been extended to the case where the global tumbling is anisotropic and where the angle between the principal axis of the global motion and the preferred orientation of the internal modes of motion is arbitrary.
|
|
|
A Multifrequency Electron Spin Resonance Study of T4 Lysozyme Dynamics
J.P. Barnes, Z. Liang, H.S. Mchaourab, J.H. Freed, and Wayne L Hubbell.
Biophys. J. 76, 3298-3306, (1999).
<doi: 10.1016/S0006-3495(99)77482-5>
PMID:
10354455
PMCID:
PMC1300299
|
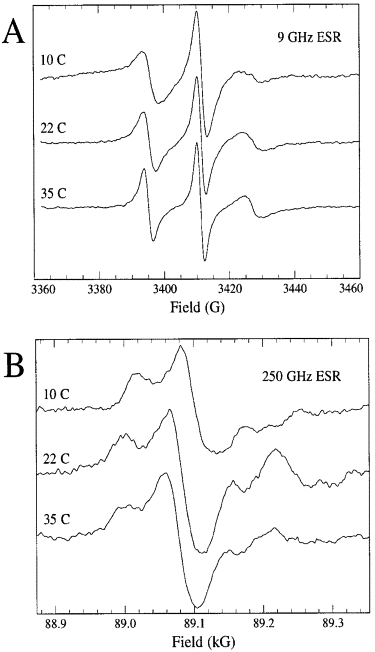
|
|
ABSTRACT: Electron spin resonance (ESR) spectroscopy at 250GHz and 9GHz is utilized to study the dynamics and local structural ordering of a nitroxide-labeled enzyme, T4 lysozyme (EC 3.2.1.17), in aqueous solution from 10°C to 35°C. Two separate derivatives, labeled at sites 44 and 69, were analyzed. The 250-GHz ESR spectra are well described by a microscopic ordering with macroscopic disordering (MOMD) model, which includes the influence of the tether connecting the probe to the protein. In the faster "time scale" of the 250-GHz ESR experiment, the overall rotational diffusion rate of the enzyme is too slow to significantly affect the spectrum, whereas for the 9-GHz ESR spectra, the overall rotational diffusion must be accounted for in the analysis. This is accomplished by using a slowly relaxing local structure model (SRLS) for the dynamics, wherein the tether motion and the overall motion are both included. In this way a simultaneous fit is successfully obtained for both the 250-GHz and 9-GHz ESR spectra. Two distinct motional/ordering modes of the probe are found for both lysozyme derivatives, indicating that the tether exists in two distinct conformations on the ESR time scale. The probe diffuses more rapidly about an axis perpendicular to its tether, which may result from fluctuations of the peptide backbone at the point of attachment of the spin probe.
|
|
|
Quasioptical Hardware for a Flexible FIR-EPR Spectrometer
K.A. Earle and J.H. Freed.
Appl. Magn. Reson. 16, 247-272 (1999).
<doi: 10.1007/BF03161937>
|
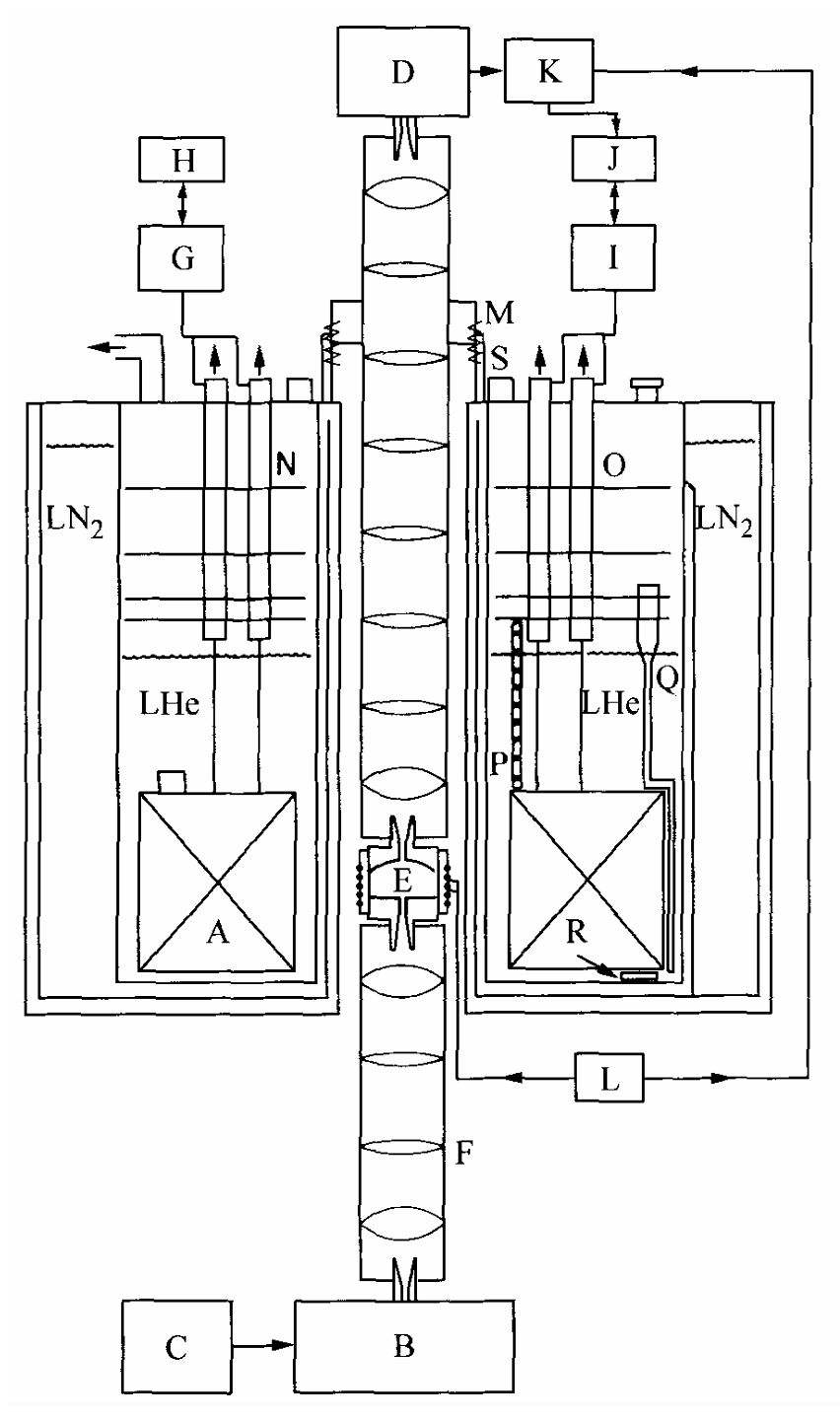
|
|
ABSTRACT: In this paper we present and summarize recent accomplishments of the Freed High Field Electron Paramagnetic Resonance group. In particular, we discuss the application of quasioptical design techniques to instrumentation problems in the far-infrared. We stress that there is no "universal spectrometer" or "universal resonator". Rather, we demonstrate with a variety of examples from the liquid and solid state that the spectrometer configuration and the resonator used should be optimized for the experiment at hand in order to achieve the ultimate in sensitivity. Quasioptical techniques and methods of analysis offer a unified framework to analyze the expected performance of a proposed spectrometer design, as well as suggest the important control parameters for optimizing the sensitivity of a given experiment as we show in the text. The flexibility of quasioptical methods will also be demonstrated with a variety of resonator designs and sample configurations.
|
|
|
A "shunt" Fabry–Perot resonator for high-frequency electron spin resonance utilizing a variable coupling scheme
J.P. Barnes and J.H. Freed.
Rev. Sci. Instrum. 69, 3022-3027 (1998).
<doi: 10.1063/1.1149050>
|
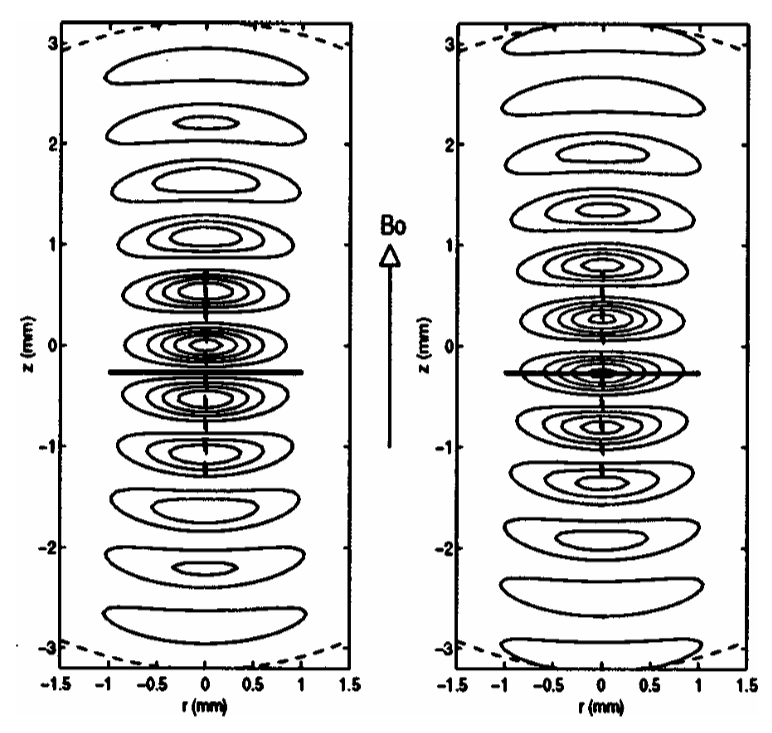
|
|
ABSTRACT: We report on the performance of a Fabry–Perot resonator for far-infrared electron spin resonance (FIR-ESR) at 250 GHz designed to accommodate a thin, disk-shaped sample that must rest with its flat surface perpendicular to the incident FIR beam. This geometry minimizes dielectric losses, making it possible to obtain FIR-ESR spectra of aqueous or lossy samples with a macroscopic ordering, at canonical values of the director tilt of 0° and 90°. The resonator also utilizes an adjustable interferometer to achieve variable coupling in the FIR regime.
|
|
|
Millimeter Wave Electron Spin Resonance Using Quasioptical Techniques
K.A. Earle, D.E. Budil, and J.H. Freed.
Adv. Magn. Optical Reson. 19, 253-323 (1996).
<doi: 10.1016/S1057-2732(96)80019-2>
|
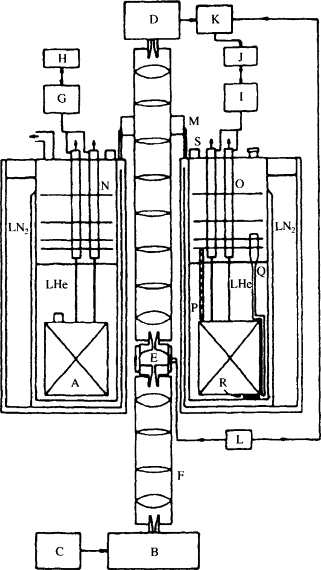
|
|
Summary: We have presented a complete analysis of the Cornell mm-wave spectrometer using quasioptical formalism with sufficient flexibility to predict the performance of a novel reflection spectrometer with variable input coupling and transmit-receive duplexing based on polarization coding and we present a practical realization of these design concepts in Earle et al. (1996b). At every stage we have chosen parameters that correspond to practical performance values and measured response.
The treatment given here is self-contained, but the references contain many useful extensions of our results and alternative treatments may deepen the reader's understanding. We have tried, where possible, to take advantage of EPR spectroscopists' knowledge of microwave circuits and analysis. As high-field ESR becomes more common, we predict that analogies from other fields will continue to be a useful method for extending the generality and utility of the method. Implementing the advanced techniques discussed in Sections IX-XI will give significant inprovements in signal-to-noise ratio as we have demonstrated elsewhere (Earle et al., 1996b).
|
|
|
Nanosecond RF Switch Driver
C. Dunnam.
Patent #5,214,315, May 25, 1993
|
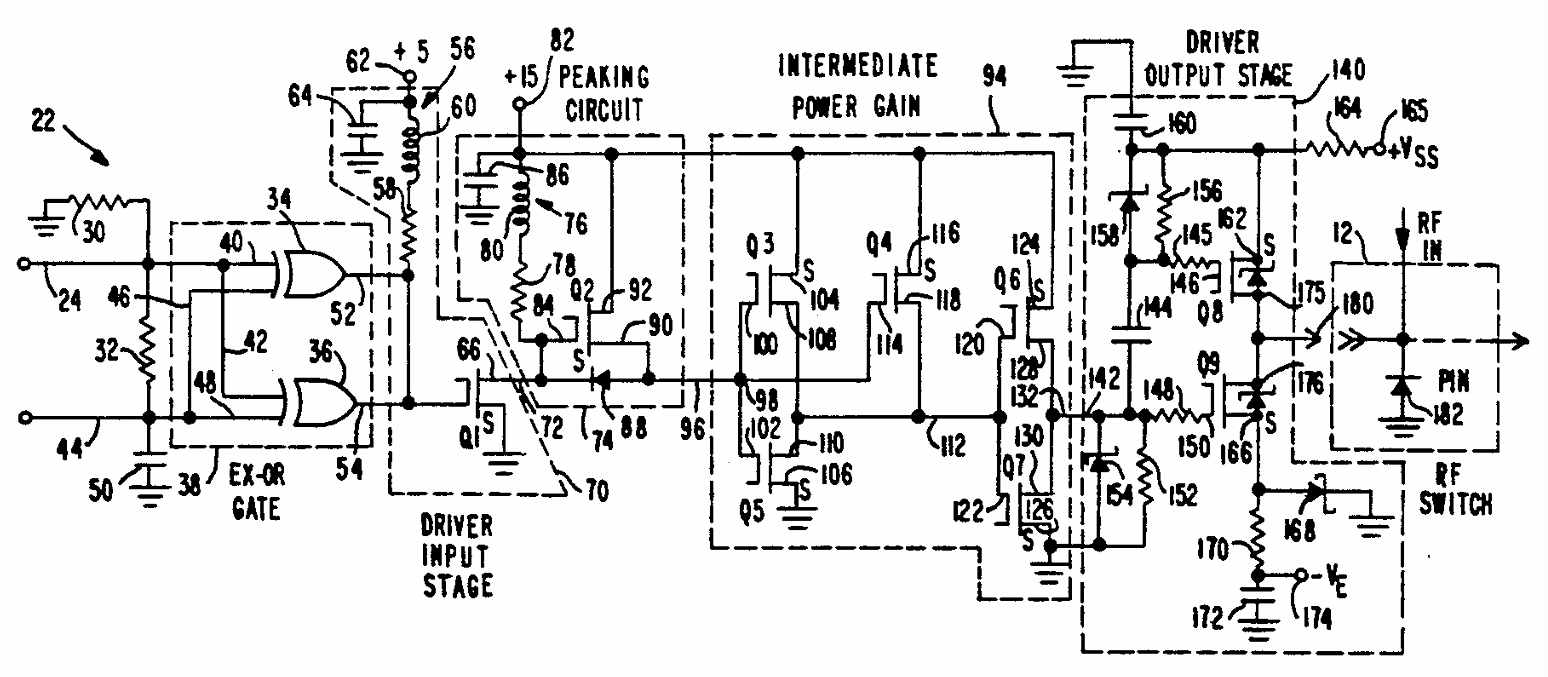
|
|
ABSTRACT: An extremely fast high-voltage driver for nanosecond-level switching of gallium arsenide PIN semiconductor devices and other reactive loads utilizes two stages of voltage level translation to permit low voltage TTL logic control signals to control the application of high voltage switching pulses to a load. The circuit utilizes an input stage responsive to TTL level signals to drive a first level translating stage which produces high voltage pulses complementary to the input pulses. The increased voltage pulses are supplied through an intermediate power gain stage to provide amplification of the power level. The amplified pulse is supplied to an output stage for additional level conversion to thereby provide high voltage switching signals to a PIN switch or other reative load.
|
|
|
Calculation of ESR Spectra and Related Fokker-Planck Forms by the use of the Lanczos Algorithm
G. Moro and J.H. Freed.
J. Chem. Phys. 74, 3757-3773 (1981).
<doi: 10.1063/1.441604>
|
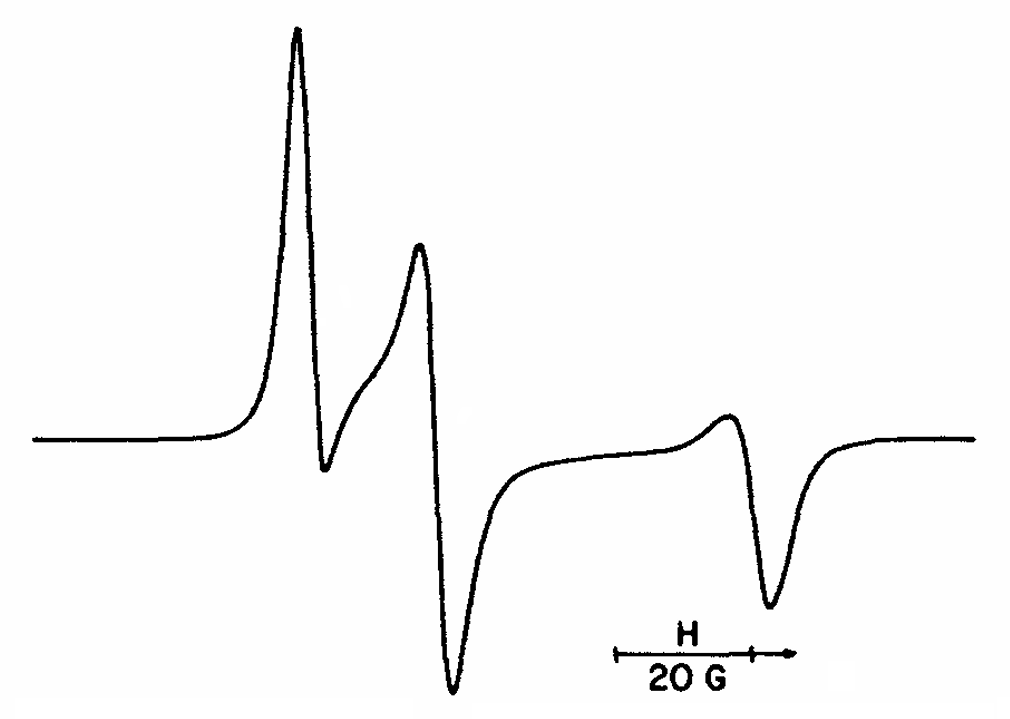
|
|
ABSTRACT: The applicability of the Lanczos algorithm in the general ESR (and NMR) line shape problem is investigated in detail. This algorithm is generalized to permit tridiagonalization of complex symmetric matrices characteristic of this problem. It is found to yield very accurate numerical solutions with at least order of magnitude reductions in computation time compared to previous methods. It is shown that this great efficiency is a function of the sparsity of the matrix structure in these problems as well as the efficiency of selecting an approximation to the optimal basis set for representing the line shape problem as distinct from actually solving for the eigenvalues. Furthermore, it is found to aid in the analysis of truncation to minimize the basis set (MTS), which becomes nontrivial in complex problems, although the efficiency of the method is not very strongly dependent upon the MTS. It is also found that typical Fokker–Planck equations arising from stochastic modeling of molecular dynamics have the property of being representable by complex–symmetric matrices that are very sparse, so calculation of associated correlation functions can be very effectively implemented by the Lanczos algorithm. It is pointed out that large problems leading to matrices of very large dimension can be efficiently handled by the Lanczos algorithm.
|
|
|
Theory of Slow Tumbling ESR Spectra for Nitroxides
J.H. Freed.
In Spin Labeling Theory and Applications, L.J. Berliner, Ed.
Academic Press: New York, 1976; Chapter 3.
|
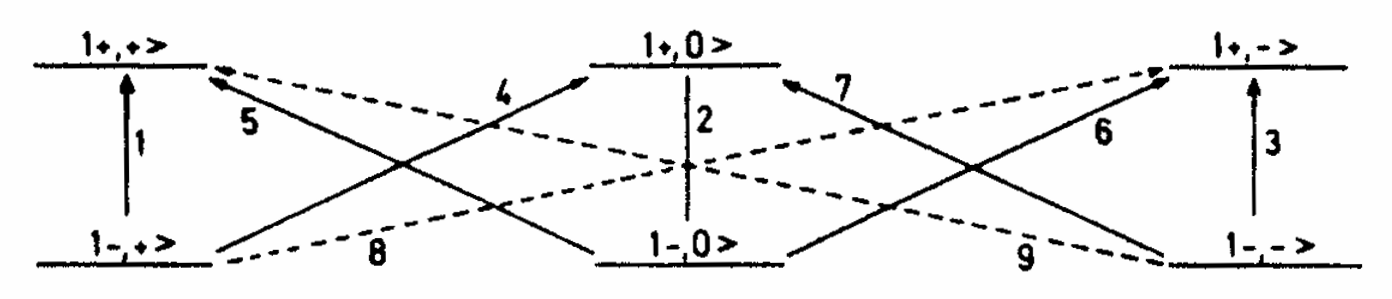
|
|
INTRODUCTION: In this chapter we develop the theory for slow tumbling in ESR spectroscopy, with specific application to nitroxide free radical spectra. The slow tumbling region is that range of rotational reorientation times for which the ESR spectrum can no longer be described as a simple superposition of Lorentzian lines, characteristic of the fast motional or motional narrowing region, for which the theory developed in the previous chapter applies. In this chapter we further take it to be the region for which the ESR spectrum still shows effects of the motion; i.e., the motion is not so slow as to yield a proper rigid-limit spectrum. For nitroxide free radicals, this usually means we are considering the range of rotational correlation times 10−9 sec ≦ τR ≦ 10−6 sec. This is an important range for nitroxide probes in viscous media or for nitroxide spin labels attached to large macromolecules.
In this region, the spectra are affected in a complicated way by both the motions and the magnetic spin interactions. As a result, a theory which can deal rigorously with describing this region must be both powerful and general. It is our objective to set forth this theory, which has been developed in the last few years, and to emphasize its general foundations. The general theory is presented in Section II.A. It is a characteristic of the slow motional region that it requires that we ask more intricate questions about the detailed nature of the molecular motions in order to properly analyze the spectra than is true for studies in the motional narrowing region. Thus in much of the remainder of Section II the dynamics of molecular motions is developed to an extent concomitant with the need to explain actual slow tumbling experiments.
|
|
© 2022
|
.svg)